
Nanoconfinement-induced orientation changes in liquid crystalline block co-oligomers†
Juyeol Kima,
Yujin Kangb,
Dong-Gue Kangc,
Hyewon Parka,
Yeongsik Kimd,
Chinedum O. Osuji c,
Hyungju Ahn*d,
Changyeon Lee*b and
Dong Ki Yoon*a
c,
Hyungju Ahn*d,
Changyeon Lee*b and
Dong Ki Yoon*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea. E-mail: nandk@kaist.ac.kr
bDepartment of Chemical Engineering, Chung-Ang University, Seoul 06974, Republic of Korea. E-mail: cylee@cau.ac.kr
cDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: cosuji@seas.upenn.edu
dPohang Accelerator Laboratory, Pohang 37673, Republic of Korea. E-mail: hyungju@postech.ac.kr
First published on 29th July 2025
Abstract
Liquid crystalline block co-oligomers (LCBCOs) are a distinct class of hybrid materials that combine the long-range structural order of block copolymers with the inherent molecular alignment capabilities of liquid crystals. Nanoconfinement using anodic aluminum oxide (AAO) templates offers a versatile platform for directing their hierarchical organization. Herein, we systematically investigate the self-assembly behavior of azobenzene-functionalized LCBCOs under nanoconfinement by varying the pore diameter (Dpore) of AAO templates from 100 to 30 nm. Grazing incidence small-angle X-ray scattering analysis reveals that different smectic mesophases—such as bilayer, interdigitated, and monolayer structures—emerge as Dpore decreases. The reduction in Dpore also induces a reorientation of LCBCO smectic layers from perpendicular to parallel alignment with respect to the surface normal. We find that these structural transitions are coupled with confinement-induced reorientation of azobenzene mesogens, which is further influenced by surface anchoring effects. Our results underscore the profound impact of nanoconfinement on the self-assembly and molecular orientation of LCBCOs, offering a promising strategy for designing next-generation anisotropic soft materials and stimuli-responsive systems.
Introduction
Precise control over self-assembly of soft matter at the nanoscale is critical for enhancing their optical,1,2 electrical,3,4 and mechanical properties,5,6 as well as for realizing novel functionalities unattainable with conventional systems. Among functional soft matter, block copolymers (BCPs) have garnered considerable attention due to their versatile functionalities stemming from diverse microphase-separated nanostructures such as lamellae, cylinders, gyroids and spheres.7–11 Despite their versatility, BCPs may be less suitable for applications that require dynamic and fast responsiveness to external stimuli. Typically high molecular weights (>20 kDa) of BCPs, while essential for microphase separation, often result in chain entanglement, thereby imposing kinetic constraints on the system.To address these limitations, liquid crystalline block co-oligomers (LCBCOs) have emerged as promising hybrid materials that combine the structural ordering of BCPs with the dynamic responsiveness of liquid crystals (LCs).12–14 Attributed to their low molecular weight (<2 kDa) and discrete block structures, LCBCOs can exhibit rapid and precise self-assembly, rendering them attractive responsive materials.15–20 Translating this promising characteristic into macroscopic functionalities requires tailoring the morphology of LCBCOs in large areas. Particularly, programming alignment of anisotropic LCBCO assemblies is crucial.
Nanoconfinement using porous anodic aluminum oxide (AAO) templates offers an effective approach to control the morphologies of LCBCOs in large areas.21–28 The highly ordered nanoporous structures of AAO can provide (i) geometric confinement and (ii) surface-mediated interactions, both inducing directed self-assembly of LCBCOs.29–32 In addition, the dense, hexagonally arranged pore arrays of AAO enable large-area uniform alignment, which is not readily achieved using conventional methods such as mechanical rubbing, surface modification, or external field-assisted alignment.33–37 While AAO has been widely used to align BCPs or LCs for enhancing anisotropic properties,38–40 the alignment behavior of LCBCOs within AAO, particularly in relation to the geometric parameters of AAO, remains unexplored.
In this study, we examine the nanoconfinement effects on an azobenzene-functionalized LCBCO (Azo-LCBCO) using AAO templates with systematically varied pore diameter (Dpore). Grazing incidence X-ray diffraction (GIXD) results demonstrate that the nanoconfinement by AAO provides access to smectic mesophases that are not observed in the bulk state and enables precise control over layer alignment. Furthermore, nanoconfinement imposes homogeneous anchoring conditions of azobenzene mesogens at the AAO interface and modulates π–π interactions within the smectic layer, collectively directing LCBCO orientation and nanoscale organization. These findings highlight the utility of AAO in enabling precise and uniform alignment of LCBCOs. They also provide key insights into how nanoconfinement can be used to tune mesophases of LCBCOs for the design of advanced functional materials.
Results and discussion
Molecular design and nanoconfinement of Azo-LCBCO within AAO nanochannels
NO2-Azo12-10-ODMS Azo-LCBCO was synthesized to investigate its orientation behavior under nanoconfinement (Fig. 1a, Scheme S1 and Fig. S1, S2, ESI†). Azo-LCBCO consists of an azobenzene-based LC block, an alkyl spacer, and a non-mesogenic oligo(dimethyl siloxane) (ODMS) block with a number-average molecular weight of ∼1 kDa. Elementary analysis suggests that the experimentally determined nitrogen content, originating from the azobenzene moieties, closely matches the theoretical values calculated for ODMS backbones with 8 or 9 repeating units. This degree of polymerization corresponds well with the Mn (1 kDa) of the ODMS–COOH precursor. Structurally similar compounds are known to self-assemble into hierarchically ordered lamellar superstructures with long-range periodicity (Fig. 1b). In this system, the ODMS segment, with its inherently low glass transition temperature, imparts high molecular mobility even at room temperature, enabling rapid self-assembly. In addition, its high dissimilarity with the Azo segment facilitates the formation of well-defined smectic layers.13 The alkyl spacer promotes microphase separation between the LC and ODMS domains, facilitating the formation of well-ordered lamellar structures. In addition, the strong electron-withdrawing NO2 end functional group reduces the energy barrier for cis-to-trans isomerization, increasing its photoresponsivity to UV light. The synthesized Azo-LCBCO exhibits a melting temperature (Tm) of 76.2 °C with a heat of fusion of 110.8 J g−1 (Fig. S2, ESI†) and shows an absorption peak at 380 nm in the UV-vis spectrum (Fig. S3, ESI†). | ||
| Fig. 1 (a) Chemical structure and schematic illustration of NO2-Azo12-10-ODMS (Azo-LCBCO). (b) Schematic illustration of the lamellar superstructure comprising smectic layers formed via self-assembly of Azo-LCBCO. (c) Schematic illustration of PEG-treated anodic aluminum oxide (AAO) templates filled with Azo-LCBCO. (d) Top-down SEM images of AAO templates with pore diameters (Dpore) of 30, 60, and 100 nm. Scale bars are 200 nm. (e) Cross-sectional SEM image of an AAO template with a Dpore of 60 nm filled with Azo-LCBCO, along with a schematic illustration of the self-assembled lamellar structures within the AAO nanochannel. The scale bar is 500 nm. | ||
The synthesized Azo-LCBCO was infiltrated into porous AAO templates containing vertically aligned nanochannels with Dpore values of 30, 60, and 100 nm and lengths of ∼5 μm (Fig. 1c and d; see the Experimental section for details). To increase the surface energy, the channel walls were chemically modified with PEG. The molten state of Azo-LCBCO was injected into the nanochannels via capillary action at 120 °C, which is above its melting temperature, followed by annealing for 90 min. Differential scanning calorimetry (DSC) analysis confirmed a decrease in the transition temperature (Fig. S6, ESI†), which was attributed to homogeneous nucleation of Azo-LCBCO within the uniformly confined geometry of the AAO template, indicating the effective influence of nanoconfinement.31 Meanwhile, Fourier transform infrared (FTIR) spectroscopy verified successful PEG surface modification and indicated negligible chemical bonding between Azo-LCBCO and the AAO interface (Fig. S7, ESI†). Residual material on the AAO surface was completely removed to isolate nanoconfined structures, and the samples were cooled to room temperature at 0.5 °C min−1. Successful infiltration into the AAO nanochannels was confirmed through cross-sectional SEM (Fig. 1e and Fig. S4, ESI†).
Self-assembly behavior of Azo-LCBCO in the bulk state
Prior to examining nanoconfined behavior, the self-assembly and orientation characteristics of Azo-LCBCO in the bulk state were analyzed (Fig. 2). The material was sandwiched between PEG-treated glass substrates, consistent with the AAO surface treatment. The polarized optical microscopy (POM) image revealed randomly oriented crystalline textures with strong birefringence, indicative of multi-domain growth without preferential alignment (Fig. 2a). AFM data further confirmed the presence of lamellar superstructures lacking directional order (Fig. 2b). The 2D small-angle X-ray scattering (SAXS) image showed an isotropic diffraction pattern and its second-order harmonic, corresponding to smectic layers formed by azobenzene moieties (q′, domain spacing ds = 4.1 nm) (Fig. 2c). While this smectic layer in the bulk state is interpreted as a bilayer, later experiments demonstrate that enhanced nanoconfinement by decreasing Dpore sizes enables access to diverse smectic mesophases of Azo-LCBCO depending on the interlayer arrangement of azobenzene mesogens (Fig. 2d). Possible configurations include bilayer (q′), tilt, interdigitated (q′′), and monolayer (q′′′) mesophases, each distinguished by their relative layer spacings (Fig. 2e).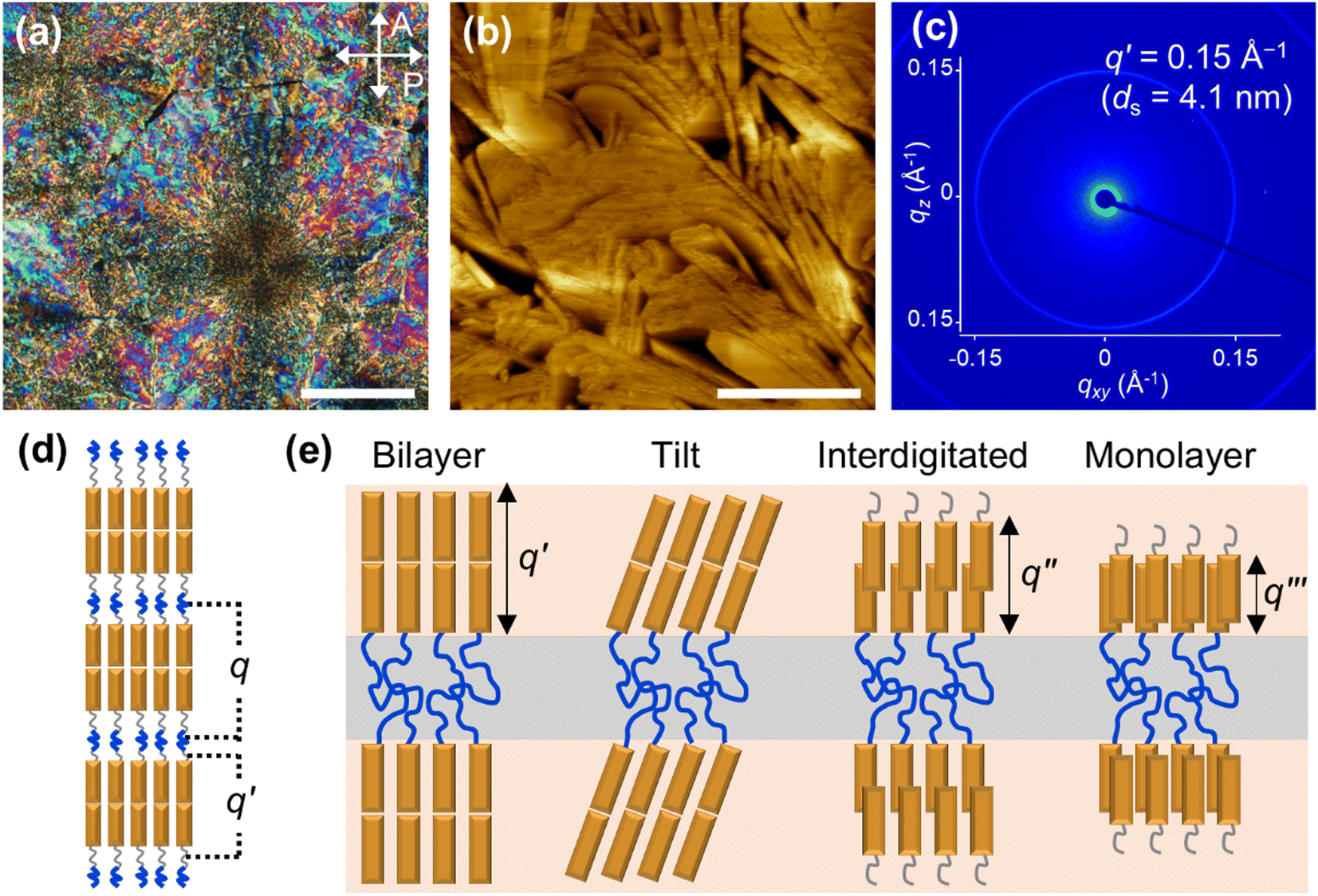 | ||
| Fig. 2 (a) POM image and (b) AFM image of Azo-LCBCO self-assembled in a PEG-treated glass sandwich cell with a 5 μm gap. Scale bars are (a) 200 μm and (b) 1 μm, respectively. (c) 2D XRD image of bulk Azo-LCBCO. (d) Schematic illustration of the self-assembly of Azo-LCBCO, where q and q′ indicate the lamellar superstructure and the smectic ordering of the Azo mesogens. (e) Schematic illustration of possible configurations of smectic mesophases. | ||
Nanoconfinement-induced self-assembly of Azo-LCBCO
The effect of nanoconfinement on the structural organization of Azo-LCBCO was investigated using GI-SAXS (Fig. 3). The third-order harmonic of the lamellar superstructure (3q) and the bilayer smectic mesophase (q′, dq′ = 4.2 nm) were consistently observed across all Dpore values, indicating stable hierarchical ordering within the AAO nanochannels (Fig. 3a–d). As Dpore decreased, the intensity of diffraction peaks corresponding to smectic mesophases increased (Fig. 3a–c). At smaller Dpore values, new diffraction peaks (q′′, dq′′ = 3.7 nm; q′′′, dq′′′ = 3.3 nm) emerged, corresponding to interdigitated and monolayer smectic mesophases, respectively. The relative intensity of these peaks increased with decreasing Dpore, indicating a structural transition driven by confinement. Mesophase ratios were quantified as the intensity fraction of each mesophase relative to the total smectic mesophase signal (Fig. 3e). While only the bilayer mesophase was present in the bulk and at Dpore = 100 nm, its fraction diminished at smaller Dpore, giving way to the other mesophases. These transitions reflect the emergence of new thermodynamically favorable configurations of Azo-LCBCO enabled by confinement.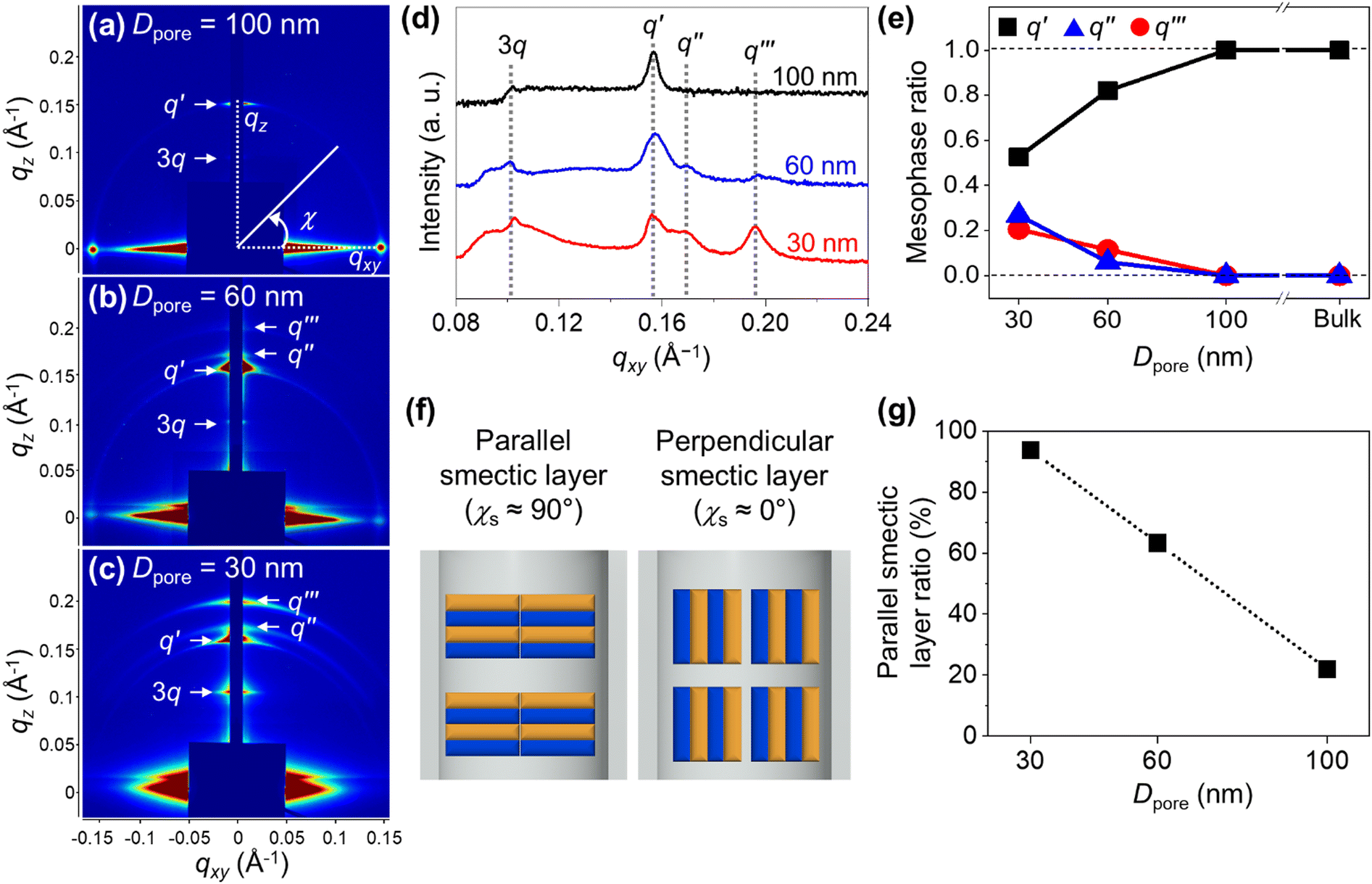 | ||
| Fig. 3 (a)–(c) 2D GI-SAXS images of Azo-LCBCO confined in AAO templates with Dpore values of (a) 100 nm, (b) 60 nm, and (c) 30 nm. (d) Line-cut profiles of the 2D GI-SAXS images of Azo-LCBCO confined in AAO templates with various Dpore values. (e) Mesophase ratio of Azo-LCBCO as a function of Dpore. (f) Schematic illustration of the orientations of smectic layers. (g) Ratio of parallel smectic layers in Azo-LCBCO as a function of Dpore. | ||
The orientation of bilayer smectic layers was assessed using azimuthal angle distributions. The azimuthal angle of the smectic layers (χs) was defined as the angle between the scattering vector q and the qxy plane, with 0° indicating the perpendicular and 90° the parallel orientation (see Fig. 3f and Fig. S8 (ESI†) for the azimuthal profile). The changes in orientation were quantified using the parallel smectic layer ratio, calculated as Apara/[Apara + Aperp], where Aperp and Apara represent the integrated areas along with χs = 3°–45° and 45°–87°, respectively (Fig. 3g). As Dpore decreased, the parallel smectic layer ratio drastically increased from 22% to 93.7%.
Mesogen orientation in nanoconfined Azo-LCBCO
The molecular orientation of azobenzene mesogens under nanoconfinement was also investigated using GI-WAXD (Fig. 4). Fig. 4a–c presents 2D GI-WAXD patterns of Azo-LCBCO confined in AAO templates with varying Dpore, exhibiting a diffraction peak from π–π interactions between azobenzene mesogens (qπ) with a corresponding spacing of 4.2 Å (Fig. 4d). A comparison with bulk WAXD data (Fig. S9, ESI†) confirms that this qπ reflection is an intrinsic feature of Azo-LCBCO, and nanoconfinement dictates the orientation of the Azo-LCBCO assemblies. | ||
| Fig. 4 (a)–(c) 2D GI-WAXD images of Azo-LCBCO confined in AAO templates with Dpore values of (a) 100 nm, (b) 60 nm, and (c) 30 nm. (d) Line-cut profiles of the 2D GI-WAXD images of Azo-LCBCO confined in AAO templates with various Dpore values. (e) Azimuthal intensity profiles at q = 1.5 Å−1 depending on Dpore. (f) Schematic illustration of the azobenzene mesogen orientation corresponding to the angular region of χm. (g) Ratio of highly tilted mesogens as a function of Dpore. | ||
Azimuthal angle distributions of the qπ peak were analyzed to evaluate mesogen orientation (Fig. 4e and Fig. S10, ESI†). At Dpore = 100 nm, the diffraction intensity was predominantly located in the high-angle region (60° ≤ χm ≤ 75°), indicating a preferential alignment of mesogens normal to the channel walls. As Dpore decreased, the intensity in this high-angle region shifted to the low-angle region (15° ≤ χm ≤ 30°), signifying reorientation parallel to the channel axis (Fig. 4f). This trend was quantified by the low χm region ratio, defined as Alow/[Alow + Ahigh], where Alow and Ahigh are the integrated areas over χm = 15°–30° and 60°–75°, respectively (Fig. 4g). As Dpore decreased, this ratio increased from 26% to 86.6%. The observed reorientation is attributed to favorable interfacial affinity between the NO2-terminated azobenzene mesogens and the PEG-modified alumina surface.
Under strong confinement, dipolar and dispersion interactions with the hydrophilic and neutrally polar PEG interface promote mesogen alignment along the channel axis. Combining these GI-WAXD results with GI-SAXS data (Fig. 3), we interpret that Azo-LCBCOs undergo a cooperative self-assembly process. Under strong confinement (i.e., in channels with a smaller Dpore), the azobenzene mesogens are homogeneously anchored at the nanochannel wall. This anchoring constraint at the molecular level is transferred to the smectic layers on a larger scale, causing them to align parallel with respect to the surface normal (as observed in Fig. 3g). Such hierarchical, cooperative assembly has been previously observed in liquid crystalline block copolymers.41–43
Effect of UV irradiation on mesogen orientation under nanoconfinement
Azo-LCBCO can exhibit photoresponsive behaviors due to the presence of azobenzene units. To evaluate the photoresponsivity of Azo-LCBCO, UV light (365 nm, 260 mW cm−2) was applied to the sample with a working distance of 8 mm (Fig. 5a). At room temperature, Azo-LCBCO in the solid state displayed birefringent textures associated with crystalline domains (Fig. 5b and c). In contrast, upon UV exposure, Azo-LCBCO exhibited an isotropic optical texture under POM (Fig. 5d and e). trans-to-cis photoisomerization of azobenzenes disrupts molecular packing in the material, resulting in UV-induced liquefaction. | ||
| Fig. 5 (a) Schematic illustration of the experimental setup for UV irradiation on the AAO template filled with Azo-LCBCO. (b) and (d) Photographs of Azo-LCBCO before and during UV irradiation. Scale bars are 10 mm. (c) and (e) POM images of Azo-LCBCO before and during UV irradiation. Scale bars are 200 μm. (f)–(h) 2D GI-WAXD images of UV-irradiated Azo-LCBCO confined in AAO templates with Dpore values of (f) 100 nm, (g) 60 nm, and (h) 30 nm. (i) Azimuthal intensity profiles at q = 1.5 Å−1 depending on Dpore. (j) Schematic illustration of azobenzene mesogen orientation upon UV irradiation depending on Dpore. | ||
To investigate the alignment behaviors of LCBCOs under UV irradiation, Azo-LCBCO melted at 120 °C was infiltrated into AAO and subsequently cooled to ambient temperature. The sample was subjected to continuous UV irradiation to maintain the cis-isomer state during alignment. After cooling, UV irradiation was ceased, allowing thermal relaxation back to the trans-azobenzene and consequent resolidification of the LCBCO. As the AAO Dpore decreased, the qπ peak shifted toward a lower χm region (Fig. 5f–h), with the intensity increasing in the 15°–30° and 150°–165° ranges (Fig. 5i). This result suggests that UV-irradiated azobenzene mesogens are preferentially aligned parallel to the narrower channel wall (Fig. 5j), which is consistent with the results without UV irradiation. These results confirm that nanoconfinement remains effective in directing mesogen alignment even under external stimuli, demonstrating its utility for tunable LCBCO alignment in dynamic environments.
Conclusions
In summary, we investigated the self-assembly behavior and alignment control of NO2-terminated Azo-LCBCO under nanoconfinement using AAO templates. GI-SAXS analysis revealed that decreasing Dpore from 100 nm to 30 nm induces the emergence of additional smectic mesophases, including interdigitated and monolayer structures, alongside the bilayer structure, highlighting enhanced structural diversity. In addition, the parallel smectic layer ratio increased from 22% to 93.7%, as further confirmed by azimuthal angle analysis of π–π stacking interactions via GI-WAXD. These results demonstrate that nanoconfinement provides precise control over both the polymorphism and molecular orientation of smectic structures in Azo-LCBCO. This work establishes a robust framework for directing hierarchical molecular organization in confined geometries, offering valuable insights for the design of nanostructured soft materials with tunable anisotropic properties.Experimental
Synthesis of Azo-LCBCO
The synthesis of Azo-LCBCO and its derivatives was carried out by modifying the procedure previously reported in the literature.16Synthesis of (E)-4-((4-nitrophenyl)diazenyl)phenol
4-Nitroaniline (5 g, 30 mmol) in 1 M HCl aqueous solution (50 ml) was heated to 50 °C to dissolve the contents, then cooled to 0 °C. A solution of sodium nitrite (3.7 g, 53.7 mmol) in 30 ml of water was added dropwise. Then, a solution of phenol (4.24 g, 45.0 mmol) and NaOH (1.80 g, 45.0 mmol) in water (50 ml) was added dropwise. After stirring for 1 h, the resulting precipitate was filtered and washed several times with water. The crude product was further purified by recrystallization from methanol (yield: 92%).Synthesis of (E)-12-(4-((4-nitrophenyl)diazenyl)phenoxy)dodecan-1-ol
To a solution of (E)-4-((4-nitrophenyl)diazenyl)phenol (5.0 g, 20.6 mmol) in anhydrous DMF (50 ml), K2CO3 (5.7 g, 41.2 mmol) was added, and the mixture was stirred under nitrogen for 30 min. 12-Bromo-1-dodecanol (7.7 g, 30.8 mmol) was added dropwise, and the reaction mixture was stirred at 80 °C for 48 h. After the reaction, the mixture was cooled to room temperature and diluted with ethyl acetate. The organic layer was washed several times with water and brine. The organic phase was separated, dried over anhydrous MgSO4, and filtered. The solvent was removed by evaporation under reduced pressure. The crude product was further purified by recrystallization from methanol (yield: 85%).Synthesis of (decanoyl chloride)-terminated oligomeric dimethylsiloxane (ODMS–COCl)
A solution of 5 g (carboxydecyl)-terminated polydimethylsiloxane (ODMS–COOH, Gelest, 1 kDa) in anhydrous dichloromethane (20 ml) was purged with nitrogen for 10 min. Oxalyl chloride (2.2 ml, 25.0 mmol) was added dropwise, followed by the addition of a catalytic amount of DMF (1 drop). The mixture was stirred at room temperature for 6 h. After the reaction, excess oxalyl chloride and solvent were evaporated out under reduced pressure. The crude ODMS–COCl was used in the next step without further purification.Synthesis of Azo-LCBCO
The resulting intermediate ODMS–COCl was redissolved in anhydrous dichloromethane (30 ml) and cooled to 0 °C. To this, (E)-12-(4-((4-nitrophenyl)diazenyl)phenoxy)dodecan-1-ol (6.5 g, 15.0 mmol) and triethylamine (2.1 ml, 15.0 mmol) were added sequentially under nitrogen. The reaction was stirred at room temperature for 24 h. After completion, the mixture was washed with 0.5 M HCl, saturated NaHCO3 and brine. The organic phase was concentrated under reduced pressure. The crude product was purified by silica gel chromatography with dichloromethane (yield: 57%).Preparation of AAO templates
High-purity aluminum foil (99.99%, Alfa Aesar) was sequentially cleaned with ethanol, acetone, and deionized water. Then, electrochemical polishing was performed in a mixed solution of perchloric acid and ethanol at 3 °C. The polished aluminum was anodized at 8 °C in 0.3 M oxalic acid to obtain Dpore values of 100 nm and 60 nm, and 0.3 M sulfuric acid for a Dpore of 30 nm. To achieve well-ordered AAO nanochannels, the anodized films were chemically etched in a mixed solution of chromic acid and phosphoric acid at 62 °C. A second anodization was conducted under the same conditions as the first. Finally, pore widening was carried out in 0.1 M phosphoric acid at 40 °C, yielding highly ordered, hexagonally packed nanochannels with precisely controlled Dpore.Preparation of Azo-LCBCO confined in AAO nanochannels
To increase the surface energy of the AAO nanochannel walls, a chemical surface treatment using polyethylene glycol (PEG) was applied to form self-assembled monolayers. Azo-LCBCO powders were deposited onto the surface of the AAO templates, followed by heating to 120 °C and annealing for 90 min to eliminate thermal history and facilitate stable infiltration into the nanochannels via capillary forces. After infiltration, excess materials on the surface were carefully removed using a cotton swab. The sample was then cooled to room temperature at a rate of 0.5 °C min−1. To prepare the UV-irradiated sample, 365 nm UV light at an intensity of 260 mW cm−2 was applied from a distance of 8 mm during both the annealing and cooling processes. For comparison, the bulk state of Azo-LCBCO was prepared by sandwiching the material between two Kapton tapes, followed by the identical annealing (120 °C for 90 min) and cooling (0.5 °C min−1) protocol.PEGylation procedure
To facilitate surface functionalization, the prepared AAO templates were first treated with O2 plasma for 15 minutes to activate hydroxyl groups on the pore walls. The templates were then immersed in a solution containing 13 ml of toluene, 20 μl of 3-[methoxy(polyethyleneoxy)6–9]propyltrimethoxysilane (PEG 6/9, Gelest), and 10 μl of acetic acid. The solution was vortexed to ensure homogeneous dispersion of PEG 6/9. The AAO templates were kept in the solution for 17 hours at room temperature, followed by sequential rinsing with toluene and ethanol to remove unreacted residues.Characterization
The chemical structures and purity of Azo-LCBCO and its intermediates were confirmed by proton nuclear magnetic resonance (1H NMR) spectroscopy (Bruker, Avance NEO 400 MHz) in deuterated chloroform (CDCl3). Chemical shifts were presented in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. To prevent signal broadening, solutions of ODMS–COOH and Azo-LCBCO were prepared at a dilute concentration of ∼1 mg ml−1. Solid-state cross-polarization/magic angle spinning (CP/MAS) 13C NMR spectra were acquired using a JNM EXZL400R 400 MHz spectrometer (JEOL, 400 MHz). Elemental analysis (C, H, N, O) was performed using a Flash 2000 CHNS/O analyzer (Thermo Fisher Scientific) to confirm the molecular composition of the synthesized compounds. Phase transition behavior was analyzed using differential scanning calorimetry (DSC, DSC 2500, TA Instruments) with a sample weight of 4.0 mg. To eliminate any previous thermal history, the sample was first heated to the isotropic phase. Cooling and subsequent heating scans were then conducted as a set using identical scan rates (10 °C min−1). Attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) analysis of each sample was conducted using a Nicolet iS50 instrument (Thermo Fisher Scientific). Atomic force microscopy (AFM; Multimode 8, Bruker) was used to analyze the surface morphology of the bulk state of Azo-LCBCO. Scanning electron microscopy (SEM) images were obtained using a Hitachi SU-8230 microscope at an accelerating voltage of 5 kV and a beam current of 7 μA. UV-vis (UV-1900i, Shimadzu) spectra were measured at a data interval of 1.0 nm and a slow scan speed. Azo-LCBCO was dissolved in chloroform at 0.0013% (w/v, 0.013 mg ml−1). Grazing incidence X-ray diffraction (GIXD) experiments were conducted at the 9A U-SAXS beamline of the Pohang Accelerator Laboratory (PAL). During the GI-SAXS experiment, the central region was masked with a lead plate to block the intense diffraction peaks originating from the AAO template. The energy of the incident X-ray beam was 11.08 keV, and the sample-to-detector distances for GI-SAXS and GI-WAXD were 2.5 m and 0.2 m, respectively.Author contributions
J. K., H. P., C. L., and D. K. Y. designed the research project, analyzed the data, and contributed to the writing of the manuscript. Y. K. and D. G. K. synthesized and characterized the Azo-LCBCO material. J. K., H. P., Y. K., and H. A. performed and analyzed the GIXD experiments. H. P. and C. O. provided critical feedback and assisted with manuscript revisions.Conflicts of interest
The authors declare no competing financial interests.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This study was supported by the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) of Korea (RS-2023-00273025, RS-2023-00212143 and RS-2023-00280532). C. O. acknowledges the support through DMR 2223705. We gratefully acknowledge the support from Pohang Accelerator Laboratory in South Korea for using the 9A beamline and their assistance with the experiments and data analysis.References
- S. Misra, L. Li, D. Zhang, J. Jian, Z. Qi, M. Fan, H.-T. Chen, X. Zhang and H. Wang, Adv. Mater., 2019, 31, 1806529 CrossRef PubMed
.
- S. Furumi, M. Kidowaki, M. Ogawa, Y. Nishiura and K. Ichimura, J. Phys. Chem. B, 2005, 109, 9245 CrossRef CAS PubMed
- G. Hyun, S. Cao, Y. Ham, D.-Y. Youn, I.-D. Kim, X. Chen and S. Jeon, ACS Nano, 2022, 16, 9762 CrossRef CAS PubMed
- P. W. Majewski, M. Gopinadhan, W.-S. Jang, J. L. Lutkenhaus and C. O. Osuji, J. Am. Chem. Soc., 2010, 132, 17516 CrossRef CAS PubMed
- E. Ma and X. Wu, Nat. Commun., 2019, 10, 5623 CrossRef CAS PubMed
- Z. Liu, M. A. Meyers, Z. Zhang and R. O. Ritchie, Prog. Mater. Sci., 2017, 88, 467 CrossRef CAS
- W. Chen and B. Wunderlich, Macromol. Chem. Phys., 1999, 200, 283 CrossRef CAS
- H. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110(1), 146 CrossRef CAS PubMed
- C. Cummins, R. Lundy, J. J. Walsh, V. Ponsinet, G. Fleury and M. A. Morris, Nano Today, 2020, 35, 100936 CrossRef CAS
- M. Walther and H. Finkelmann, Liq. Cryst., 1997, 22, 655 CrossRef CAS
- K. Masutani, Y. Yoshioka, Y. Kimura and C. W. Lee, Macromol. Res., 2024, 32, 631 CrossRef CAS
- K. Nickmans, J. N. Murphy, B. Waal, P. Leclère, J. Doise, R. Gronheid, D. J. Broer and A. P. H. J. Schenning, Adv. Mater., 2016, 28, 10068 CrossRef CAS PubMed
- R. H. Zha, B. F. M. de Waal, M. Lutz, A. J. P. Teunissen and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 5693 CrossRef CAS PubMed
- R. J. Mandle, M. P. Stevens and J. W. Goodby, Liq. Cryst., 2017, 44, 2046–2059 CrossRef CAS
- B. Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, J. Polym. Sci., 2021, 59, 373 CrossRef
- C. Lee, D. Ndaya, R. Bosire, N. K. Kim, R. M. Kasi and C. O. Osuji, J. Am. Chem. Soc., 2022, 144, 390 CrossRef CAS PubMed
- B. Genabeek, B. F. M. Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138(12), 4210 CrossRef PubMed
- P. Bohn, M. Frölich, D. Hahn, R. V. Schneider and M. A. R. Meier, Macromol. Chem. Phys., 2025, 226, 2400396 CrossRef CAS
- K. Chen, C. Lee, C.-Y. Chen, T. Satoh, T. Isono and H.-L. Chen, Giant, 2024, 19, 100308 CrossRef CAS
- C. Lee and C. O. Osuji, ACS Macro Lett., 2021, 10(7), 945 CrossRef CAS PubMed
- J.-B. Park, Y.-S. Kim, S.-K. Kim and H.-S. Lee, Bull. Korean Chem. Soc., 2004, 25, 563 CrossRef CAS
- T. Iseki, M. N. Biutty, C. H. Park and S. I. Yoo, Macromol. Res., 2023, 31, 223 CrossRef CAS
- M. Lee, S.-H. Yoon and Y. Kim, J. Inf. Disp., 2007, 8, 19 CrossRef
- C. Mijangos, R. Hernández and J. Martín, Prog. Polym. Sci., 2016, 54–55, 148 CrossRef CAS
- W. Lee and S.-J. Park, Chem. Rev., 2014, 114, 7487 CrossRef CAS PubMed
- A. Ruiz-Clavijo, O. Caballero-Calero and M. Martín-González, Nanoscale, 2021, 13, 2227 RSC
- S. H. Ryu and D. K. Yoon, ACS Appl. Mater. Interfaces, 2017, 9, 25057 CrossRef CAS PubMed
- J. Ko, R. Berger, H. Lee, H. Yoon, J. Cho and K. Char, Chem. Soc. Rev., 2021, 50, 3585 RSC
- J. S. Kim, Y. Park, D. Y. Lee, J. H. Lee, J. H. Park, J. K. Kim and K. Cho, Adv. Funct. Mater., 2010, 20, 540 CrossRef CAS
- R. M. Michell, I. Blaszczyk-Lezak, C. Mijangos and A. J. Müller, J. Polym. Sci., Part B, 2014, 52, 1179 CrossRef CAS
- Y. Guan, G. Liu, P. Gao, L. Li, G. Ding and D. Wang, ACS Macro Lett., 2013, 2(3), 181 CrossRef CAS PubMed
- H. Wu, W. Wang, Y. Huang and Z. Su, Macromol. Rapid Commun., 2009, 30, 194 CrossRef CAS PubMed
- A. Santos, T. Kumeria and D. Losic, Trends Anal. Chem., 2013, 44, 25 CrossRef CAS
- S. Liu, J. Tian and W. Zhang, Nanotechnology, 2021, 32, 222001 CrossRef CAS PubMed
- H. Park, H. Park, H. Ahn, W. Lee, J. Park, B. J. Kim and D. K. Yoon, Chem. Mater., 2023, 35, 1355 CrossRef CAS
- M. Seo and J. H. Bae, Bull. Korean Chem. Soc., 2024, 45(12), 993 CrossRef CAS
- H. Seong and D. Lee, Bull. Korean Chem. Soc., 2024, 45(5), 435 CrossRef CAS
- R. You, W. Park, E. Carlson, S. H. Ryu, M. J. Shin, E. Guzman, H. Ahn, T. J. Shin, D. M. Walba, N. A. Clark and D. K. Yoon, Liq. Cryst., 2019, 46, 316 CrossRef CAS
- L. Foley, W. Park, M. Yang, E. Carlson, E. Korblova, D. K. Yoon and D. M. Walba, Chem. – Eur. J., 2019, 25, 7438 CrossRef CAS PubMed
- W. Park, B. Feringán, M. Yang, S. H. Ryu, H. Ahn, T. J. Shin, T. Sierra, R. Giménez and D. K. Yoon, Chem. Phys. Chem., 2019, 20, 890 CrossRef CAS PubMed
- H. Yu, T. Iyoda and T. Ikeda, J. Am. Chem. Soc., 2006, 128, 11010 CrossRef CAS PubMed
- E. Verploegen, T. Zhang, Y. S. Jung, C. Ross and P. T. Hammond, Nano Lett., 2008, 8, 3434 CrossRef CAS PubMed
- C. Lee, D. Ndaya, R. Bosire, U. R. Gabinet, J. Sun, P. Gopalan, R. M. Kasi and C. O. Osuji, Macromolecules, 2021, 54, 3223 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01674f |
| This journal is © The Royal Society of Chemistry 2025 |