
Multiscale simulation study on the mechanical, electrical, and thermal properties of ZnSb semiconductor
Shihui Chena,
Hanwen Yanga,
Guang Yanga,
Minghui Chenga,
Zipei Zhanga,
Zhangfeng Wu*b and
Jianping Lin *a
*a
aFujian Provincial Key Laboratory of Functional Materials and Applications, School of Materials School of Materials Science and Engineering, Xiamen University of Technology, Xiamen 361024, China. E-mail: jplin@xmut.edu.cn
bSchool of Environmental Science and Engineering, Xiamen University of Technology, Xiamen 361021, China. E-mail: wuzhangfeng@xmut.edu.cn
First published on 29th July 2025
Abstract
This study presents a comprehensive investigation of the multifunctional properties of ZnSb semiconductors through integrated multiscale simulations. A highly accurate interatomic potential function for ZnSb was constructed and validated using density functional theory (DFT) and deep potential (DP) methods within the temperature range of 300 K to 800 K. Molecular dynamics (MD) simulations revealed the mechanical behavior of ZnSb along different crystallographic axes. The a-axis exhibited significant plastic deformation with a fracture strain of 32%, while the b-axis and c-axis demonstrated brittle fracture characteristics. As the temperature increased from 300 K to 700 K, both the elastic modulus and ultimate strength decreased significantly, indicating the detrimental effect of high temperatures on its mechanical properties. Simulations of thermoelectric performance showed that optimizing carrier concentration can significantly improve the power factor (PF). Electronic thermal conductivity (κe) increases with carrier concentration and temperature, but the Seebeck coefficient performs better at lower carrier concentrations. Thermal transport analysis revealed that the lattice thermal conductivity of ZnSb initially decreases and then increases with rising temperature, with the contribution of Sb–Sb bonds to thermal conductivity exceeding 50%. This study provides a theoretical foundation for the application of ZnSb materials in thermoelectric conversion and high-temperature devices, and highlights the key parameters for performance optimization.
1. Introduction
In recent years, energy issues have become one of the major challenges facing modern society.1,2 Semiconductor materials have garnered significant attention in the fields of energy conversion and electronic devices due to their excellent properties. Among these materials, ZnSb-based materials are considered promising candidates for mid-temperature thermoelectric converters due to their low thermal conductivity and good electrical conductivity.3–5 β-Zn4Sb3 exhibits the best thermoelectric performance among reported ZnSb-based materials. Studies reveal that its unique interstitial Zn atom diffusion behavior, strong phonon anharmonicity, and continuous phase transitions are the key mechanisms responsible for its low thermal conductivity.6,7 These findings provide important insights into understanding the thermal transport behavior of semiconductors with complex atomic arrangements and dynamic disorder. ZnSb belongs to the orthorhombic crystal system, and its mechanical and thermal transport behavior under complex temperature conditions remains unclear.8,9 Shaver et al.10 confirmed through single-crystal experiments that ZnSb exhibits significant electrical conductivity anisotropy but weak thermal conductivity anisotropy. In addition, the maximum thermoelectric figure of merit (Z = 0.74 × 10−3 K−1) can be obtained along the c-axis of single-crystal ZnSb. Zhao et al.11 used the all-electron LACO-DFT method to obtain an indirect band gap consistent with experiments, clarifying that the valence band maximum is located near the X point between Γ–X (X′ point), and the conduction band minimum is located near the Z point between Z–Γ (Z′ point). Although previous studies have explored the thermoelectric figure of merit (ZT) and crystal stability of ZnSb through experimental methods,12–14 there are still gaps in the systematic analysis of its mechanical, electrical transport, and thermal transport properties. In particular, the degradation mechanism of material performance and optimization strategies under high-temperature conditions need further investigation.The mechanical properties and thermal stability of ZnSb are crucial for its application in devices. Specifically, the mechanical properties of ZnSb vary significantly due to the anisotropy of its crystal structure.15,16 Guan et al.17 found that the Young's modulus of unstrained ZnSb along the zigzag direction is much higher than that along the armchair direction, indicating strong mechanical anisotropy. The electrical transport properties of ZnSb have also attracted considerable attention.18–20 Ostovari et al.21 found that the electrical conductivity of ZnSb increases with temperature, while the Seebeck coefficient decreases with increasing carrier concentration. Karthikeyan et al.22 used first-principles (FP) calculations to investigate the effect of carrier concentration on the thermoelectric properties of ZnSb and suggested that optimizing the carrier concentration could significantly improve the power factor (PF). The low thermal conductivity of ZnSb is advantageous for improving its thermoelectric performance. Alloying, nanostructuring, and defect engineering are key areas of current research for synergistically modulating thermal conductivity.23–25 Theja et al.26 introduced a second-phase material, γ-Al2O3, via nanocomposite technology to enhance the thermoelectric performance of ZnSb. Furthermore, Fu et al.27 prepared titanium-doped ZnSb nanocomposite thin films and improved the 10-year data retention capability of ZnSb from 103 °C to 120 °C. Studies have demonstrated that the microstructure of ZnSb has a significant impact on its thermal transport properties.28 Jia et al.29 showed that the polycrystalline structure and grain size of ZnSb significantly affect its thermoelectric performance. However, future research needs to integrate in situ characterization with multi-scale simulations.30–32 This approach will clarify two critical aspects: the electron–phonon coupling mechanism under high-temperature conditions and the influence of microscopic defects on thermal transport dynamics.33 This study employs multiscale simulation methods to systematically investigate the mechanical, electrical transport, and thermal properties of ZnSb semiconductors and their temperature dependence. First, based on DFT-optimized lattice parameters and combined with deep potential (DP), a high-precision interatomic potential function is generated, and molecular dynamics (MD) simulations are used to reveal the tensile fracture behavior of the material along different crystallographic axes. Second, Boltzmann transport theory is used to analyze the effect of carrier concentration on the Seebeck coefficient and PF. Finally, thermal conductivity is calculated using the Green–Kubo method, and the contributions of Sb and Zn atomic group to heat transport are analyzed. The research results not only provide a theoretical basis for the application of ZnSb in thermoelectric devices but also offer new insights for material design under multi-physics field coupling.
2. Computational method
We first performed FP calculations based on DFT using the existing ZnSb unit cell structure to optimize the lattice constants and atomic positions. Next, we employed the deep potential generator (DP-GEN) tool, along with VASP software, to calculate the system's energy and atomic forces under various configurations, temperatures, and pressures. To acquire sufficient training data, we performed atomic perturbations and sampling under different states on the ZnSb unit cell [Fig. 1(a)], generating a configuration dataset with rich information. Subsequently, we utilized DeepMD-kit to train a neural network on these datasets, gradually optimizing the potential energy function. The final ZnSb potential function model generated can accurately describe the interaction between Zn and Sb atoms under various conditions.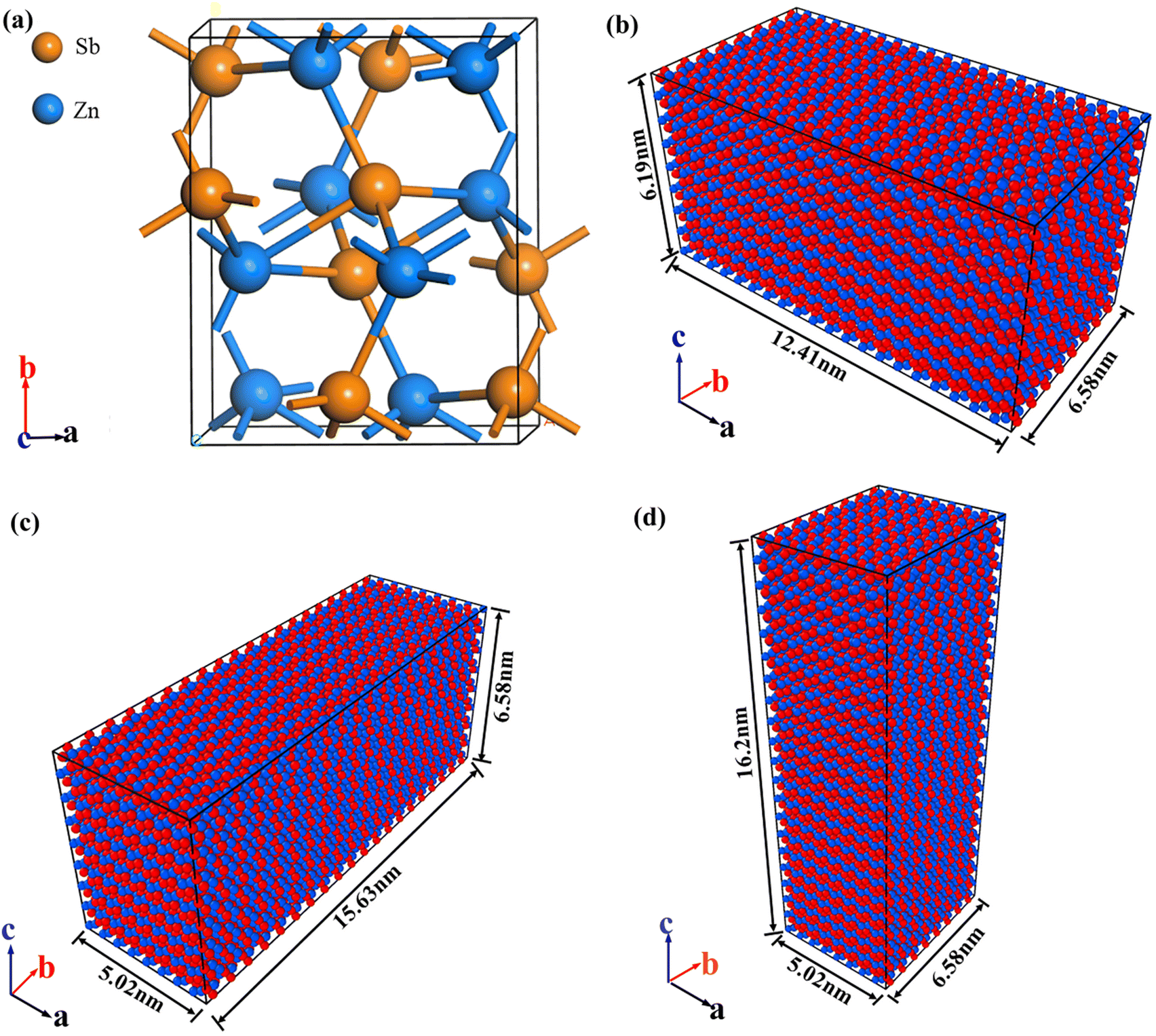 | ||
| Fig. 1 (a) Unit cell of ZnSb; uniaxial tensile models of ZnSb (b) along the a-axis; (c) along the b-axis; (d) along the c-axis. | ||
After obtaining the ZnSb potential function, we constructed three bulk models [as shown in Fig. 1(b)–(d)] for MD simulations. These models were built by periodically stacking the optimized ZnSb unit cell along the a, b, and c directions. To simulate the infinite crystal behavior of the actual material and avoid boundary effects, we applied periodic boundary conditions (PBCs) in the a, b, and c directions. To study the mechanical properties of the material in different directions, we constructed uniaxial tensile models along the a-, b-, and c-axes, respectively. In the tensile simulations along each direction, tensile stress was gradually applied by controlling the strain in the corresponding axial direction. The PBCs were maintained in the other directions to study the stress–strain behavior of ZnSb.
We also combined Boltzmann transport theory with FP calculations to systematically simulate the thermoelectric properties of ZnSb materials. First, we obtained the electronic structure through DFT calculations. Then, based on these results, we applied the Boltzmann transport equation to calculate the electrical conductivity, carrier concentrations, and thermoelectric properties of the material at different temperatures. Finally, we calculated the HCACF through MD simulations and obtained the thermal conductivity of ZnSb at different temperatures by integrating the HCACF over time and combining it with the Green–Kubo method.
3. Results and discussion
Fig. 2(a)–(c) show the radial distribution functions (RDFs) of Zn–Zn, Zn–Sb, and Sb–Sb at different temperatures. The RDFs of Zn–Sb and Sb–Sb remain stable between 300 K and 800 K. In contrast, the RDFs of Zn–Zn exhibit fluctuations at elevated temperatures, indicating increasingly dynamic Zn–Zn interactions. Fig. 2(d) shows the temperature dependence of the potential energy curve. The potential energy increases linearly with temperature, with a distinct turning point at 819 K (marked by the red dashed line), corresponding to the melting point of ZnSb. This validates the accuracy of the potential function. Fig. 2(e) shows the temperature-dependent bond lengths. Zn–Sb and Sb–Sb bond lengths remain stable, whereas the Zn–Zn bond undergoes significant elongation at 400 K and 600 K. Concurrently, the ZnSb unit cell volume expands markedly at these temperatures [Fig. 2(f)], demonstrating a direct correlation between volume expansion and Zn–Zn bond lengthening. | ||
| Fig. 2 (a) Radial distribution functions (RDFs) of Zn–Zn at varying temperatures; (b) RDFs of Zn–Sb at varying temperatures; (c) RDFs of Sb–Sb at varying temperatures; (d) temperature-dependent potential energy of ZnSb; (e) temperature-dependent bond length of ZnSb; (f) temperature-dependent unit cell volume of ZnSb. | ||
Fig. 3(a)–(c) show the deformation and fracture of ZnSb under tensile strain along the a-, b-, and c-axes. Under a-axis tension, the lattice maintains order up to 25% strain, fracturing at 32%. In contrast, b- and c-axes tensions induce lattice defects at 10% strain, leading to fracture at 14% and 12% strain, respectively. Fig. 3(d) compares the stress–strain curves for different crystallographic axes. Along the b- and c-axes, limited elastic deformation precedes rapid brittle fracture. Along the a-axis, prolonged plastic deformation follows elastic deformation before final fracture.
 | ||
| Fig. 3 (a)–(c) Structural evolution of ZnSb under uniaxial tension along the a-, b-, and c-axes, respectively, showing deformation and fracture initiation; (d) comparative stress–strain curves for the a-, b-, and c-axes at 300 K. | ||
The significant anisotropy in the mechanical behavior of ZnSb is closely related to its orthorhombic crystal structure (Pnma space group). The superior plastic deformation capability observed along the a-axis suggests the presence of relatively weak atomic bonding or easily activated specific slip systems. These systems allow dislocations to continuously slip under higher stress, resulting in a large degree of shear deformation without immediate fracture. In contrast, the b-axis exhibits high elastic modulus and brittle fracture characteristics. This indicates the likely presence of a strong covalent bond network along this direction. This network hinders dislocation nucleation and movement, leading to rapid cleavage fracture when the material reaches its theoretical strength limit. The brittle behavior along the c-axis is likely a result of its bonding strength and slip characteristics being intermediate between those of the a- and b-axes.
Fig. 4(a) shows the temperature-dependent lattice parameters of ZnSb. As temperature increases from 300 K to 700 K, parameters a, b, and c expand approximately linearly, indicating that the crystal structure expands with increasing temperature. The simulation results (black squares) align closely with those of A. Fisher (red circles). The increase in parameter a is the most significant, while that of c is the smallest, reflecting structural anisotropy. Fig. 4(b) shows the temperature dependence of elastic moduli. The moduli in all three directions decrease with increasing temperature, indicating material softening at elevated temperatures. The elastic modulus in the b-axis direction is the highest, exhibiting strong rigidity, whereas that in the a-axis direction is the lowest, suggesting easier deformation. Fig. 4(c) shows the temperature-dependent ultimate strength. The strength decreases in all three directions with increasing temperature. The b-axis exhibits the highest ultimate strength, whereas the a-axis shows the lowest, highlighting superior load-bearing capacity along the b-axis direction. Fig. 4(d) shows the trend of fracture strain with temperature. While the temperature increase induces only marginal variations in fracture strain overall, the a-axis orientation exhibits the highest fracture strain values, demonstrating superior ductility. In contrast, the c-axis direction shows the lowest fracture strain measurements, characteristic of more brittle fracture behavior. Overall, the degradation of mechanical properties (e.g., strength and stiffness) with temperature implies inherent limitations for ZnSb in high-temperature applications, necessitating targeted performance optimization.
 | ||
| Fig. 4 (a) Lattice parameters of ZnSb along the a-, b-, and c-axes at varying temperatures; (b) elastic moduli along the a-, b-, and c-axes at varying temperatures; (c) ultimate strength of ZnSb along the a-, b-, and c-axes at varying temperatures; (d) fracture strain along the a-, b-, and c-axes at varying temperatures. | ||
The decrease in elastic modulus and ultimate tensile strength with increasing temperature is a result of intensified lattice thermal vibrations. As temperature rises, the kinetic energy of atoms increases, leading to a larger average interatomic spacing. This increase in interatomic spacing directly weakens the bonding strength between atoms. Microscopically, the elastic modulus reflects the curvature of the interatomic force curve near the equilibrium position. Weakened bonding inevitably leads to a decrease in the elastic modulus. The decrease in ultimate tensile strength reduces the material's ability to resist permanent deformation and fracture. This decline stems from both the weakening of bond strength and thermally activated processes that promote dislocation nucleation and slip. Notably, lattice expansion is most significant along the a-axis. This further confirms that bonding in this direction is most sensitive to temperature changes.
Fig. 5(a) shows the variation of electrical conductivity with carrier concentration. Electrical conductivity rises sharply with increasing carrier concentration, particularly at high concentrations (>1021 cm−3), where the enhancement becomes pronounced, reflecting superior electrical conductivity in ZnSb under such conditions. While the temperature effect on conductivity is modest, a slight increase is still observed at elevated temperatures. Electrical conductivity is proportional to both carrier concentration and mobility (μ). As temperature increases, phonon scattering intensifies, leading to a decrease in carrier mobility. The decrease in mobility and the increase in carrier concentration have counteracting effects. This is why the effect of temperature increase on conductivity is relatively small. Fig. 5(b) illustrates the carrier-concentration dependence of κe. Similar to electrical conductivity, κe increases with carrier concentration, especially at high temperatures (e.g., 700 K), where electronic contributions dominate heat transport. This trend highlights the growing role of electrons in thermal conduction at elevated temperatures. This phenomenon is consistent with the Wiedemann–Franz law. This law states that the electronic contribution to thermal conductivity is proportional to electrical conductivity and temperature. Therefore, the increase in κe/τ is a result of the combined effect of the increase in σ/τ and the rise in temperature. Fig. 5(c) shows the trend of the Seebeck coefficient with carrier concentration. At low carrier concentrations, the Seebeck coefficient remains high but drops rapidly with increasing carrier concentration. This is because high carrier concentrations reduce the thermoelectric potential difference of the material, leading to a decrease in the Seebeck coefficient. Notably, the Seebeck coefficient retains higher values at lower temperatures (e.g., 300 K), suggesting ZnSb's enhanced thermoelectric potential under low temperature and low carrier concentration conditions. Fig. 5(d) shows the variation of the PF with carrier concentration. The power factor exhibits three peaks as the carrier concentration increases. The maximum peak occurs at a carrier concentration of about 1021 cm−3. Increasing temperature significantly increased the peak of the PF, especially at the highest value at 700 K, indicative of improved thermoelectric conversion efficiency at high temperatures. These results demonstrate that the thermoelectric performance of ZnSb can be effectively optimized by adjusting the carrier concentration. Strategic adjustment of carrier concentration under varying temperature conditions is critical for maximizing the material's thermoelectric efficiency.
 | ||
| Fig. 5 Variation of ZnSb's electrical transport properties with carrier concentration at different temperatures: (a) electrical conductivity/relaxation time; (b) electronic thermal conductivity/relaxation time; (c) Seebeck coefficient; (d) power factor (S2σ)/relaxation time. | ||
Fig. 6(a) shows the HCACF of a ZnSb single crystal bulk as a function of time at 300 K. The HCACF decays rapidly with time and tends to zero, indicating that the system reaches thermal equilibrium quickly. Fig. 6(b) shows the variation of thermal conductivity with time at 300 K. It indicates that the calculated thermal conductivity is reliable within this time range. By smoothing the curve, the final calculated thermal conductivity stabilizes at approximately 1.6 W m−1 K−1. Fig. 6(c) shows the trend of lattice thermal conductivity of ZnSb with temperature. The lattice thermal conductivity of ZnSb gradually decreases from about 1.6 W m−1 K−1 at 300 K to about 0.8 W m−1 K−1 at 500 K, which is related to the enhanced lattice vibration and intensified phonon scattering with increasing temperature. Subsequently, the lattice thermal conductivity increases to about 1.0 W (m K)−1 at 700 K, which is attributed to the increase in carrier concentration caused by intrinsic excitation. The simulated lattice thermal conductivity closely matches the experimentally measured value for ZnSb. The numerical deviation from room temperature to 500 K is less than 12.5%, and the simulation and experimental results show a minor difference above 650 K. This minor discrepancy could potentially be attributable to a combination of material-related factors (e.g., impurity content or defect density) and experimental uncertainties in the original data acquisition. Fig. 6(d) shows the contribution of different atomic groups to the total lattice thermal conductivity of ZnSb. The results show that Sb–Sb bond contribute the most to the thermal conductivity, accounting for 50–60%, indicating that it plays a dominant role in heat conduction. In contrast, the contribution of Zn–Zn bond is smaller, around 20–30%, while the Zn–Sb interaction contributes the remaining 10–20%. As the temperature increases, the contribution ratio of each atomic group remain relatively stable, indicating that the heat conduction mechanism of ZnSb does not change significantly with temperature.
 | ||
| Fig. 6 (a) Time-dependent heat current autocorrelation function (HCACF) of ZnSb single crystal at 300 K; (b) time-dependent thermal conductivity of ZnSb single crystal at 300 K; (c) comparison between molecular dynamics simulation results and experimental results for the lattice thermal conductivity of ZnSb; (d) proportional contributions of each pair of atom types to the total lattice thermal conductivity of ZnSb. | ||
4. Conclusions
This study provides a systematic analysis of the mechanical, electrical, and thermal properties of the ZnSb semiconductor and their temperature dependence, using FP calculations, MD simulations, and Boltzmann transport theory. The results reveal that ZnSb exhibits significant anisotropic mechanical behavior, with superior plastic deformation ability along the a-axis compared to the b- and c-axes. However, high temperatures cause a decrease in both elastic modulus and ultimate strength, which limits its application in high-temperature environments. In terms of electrical transport properties, carrier concentration is a key factor influencing thermoelectric performance. The PF reaches its optimal value when a moderate carrier concentration (1020–1022 cm−3) is combined with high-temperature (700 K), offering valuable insights for improving thermoelectric conversion efficiency. Regarding thermal transport, Sb atoms dominate the thermal conductivity, which initially decreases and then increases with rising temperature. In the low-temperature regime, phonon scattering is the main contributor, while at higher temperatures, carrier contribution becomes more significant. The simulation results align well with experimental data, confirming the reliability of the model. Overall, this study deepens the understanding of ZnSb and provides theoretical guidance for the design and engineering of new semiconductor materials.Conflicts of interest
The authors declare that they have no conflict of interest.Data availability
The data supporting this article, including mechanical analysis, thermal transport analysis, and electrical transport analysis of ZnSb semiconductor, are available from the corresponding author upon reasonable request. Upon acceptance, the data will be deposited in a suitable public database and shared with the appropriate accession details.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No. 52073240), Natural Science Foundation of Fujian Province (Grant no. 2024J011209).References
- W. Xu, Q. Liu, X. Zhou, J. Lin, S. Lin, M. Lu and J. Lin, Effects of stresses on the thermoelectric properties of In4Se3, J. Mater. Chem. C, 2024, 12(14), 5062–5072, 10.1039/D3TC04092E
.
- S. Chaudhuri, A. Bhattacharya, A. K. Das, G. P. Das and B. N. Dev, Strain driven anomalous anisotropic enhancement in the thermoelectric performance of monolayer MoS2, Appl. Surf. Sci., 2023, 626, 157139, DOI:10.1016/j.apsusc.2023.157139
- G. J. Snyder and E. S. Toberer, Complex Thermoelectric Materials, Nat. Mater., 2008, 7(2), 105–114, DOI:10.1038/nmat2090
- S. Yang, T. Deng, P. Qiu, T. Xing, J. Cheng, Z. Jin, P. Li, X. Shi and L. Chen, High-Performance and Stable (Ag, Cd)-Containing ZnSb Thermoelectric Compounds, ACS Appl. Mater. Interfaces, 2022, 14(23), 26662–26670, DOI:10.1021/acsami.2c03304
- I. Paulraj, V. Lourdhusamy, Z.-R. Yang, C.-H. Wang and C.-J. Liu, Enhanced thermoelectric properties of porous hybrid ZnSb/EG-treated PEDOT:PSS composites, J. Power Sources, 2023, 572, 233096, DOI:10.1016/j.jpowsour.2023.233096
- J. Lin, X. Li, G. Qiao, Z. Wang, J. Carrete, Y. Ren, L. Ma, Y. Fei, B. Yang, L. Lei and J. Li, Unexpected high-temperature stability of β-Zn4Sb3 opens the door to enhanced thermoelectric performance, J. Am. Chem. Soc., 2014, 136(4), 1497–1504, DOI:10.1021/ja410605f
- X. Li, J. Carrete, J. Lin, G. Qiao and Z. Wang, Atomistic origin of glass-like Zn4Sb3 thermal conductivity, Appl. Phys. Lett., 2013, 103(10), 103902, DOI:10.1063/1.4820247
- Z. Guo, Y. Liu, S. Li, X. Zhang and G. Liu, Two-dimensional antiferromagnetic type-II Weyl fermions in monolayer ZnSb and type-I Weyl fermions and topological insulator in bilayer ZnSb, Comput. Mater. Sci., 2024, 231, 112540, DOI:10.1016/j.commatsci.2023.112540
- L. Hnuna, E. Haidar, B. Djamel, C. Stampfl, S. Mohammed and Z. Pachuau, First-principles study of optical and thermoelectric properties of Zn3As2 and ZnSb, Nano Sel., 2023, 4(9–10), 551–558, DOI:10.1002/nano.202300074
- P. J. Shaver and J. Blair, Thermal and Electronic Transport Properties of p-Type ZnSb, Phys. Rev., 1966, 141(2), 649, DOI:10.1103/PhysRev.141.649
- G.-L. Zhao, F. Gao and D. Bagayoko, Reliable density functional calculations for the electronic structure of thermoelectric material ZnSb, AIP Adv., 2018, 8(10), 105211, DOI:10.1063/1.5051346
- A. Fischer, E. W. Scheidt1, W. Scherer, D. E. Benson, Y. Wu, D. Eklöf and U. Häussermann, Thermal and vibrational properties of thermoelectric ZnSb: Exploring the origin of low thermal conductivity, Phys. Rev. B, 2015, 91(22), 224309, DOI:10.1103/PhysRevB.91.224309
- P. Dharmaiah, M. Heo, C. Nagarjuna, S.-J. Jung, S. O. Won, K. H. Lee, S. K. Kim, J.-S. Kim, B. Ahn, H.-S. Kim and S.-H. Baek, Enhancement of thermoelectric properties in p-type ZnSb alloys through Cu-doping, J. Alloys Compd., 2024, 1004, 175739, DOI:10.1016/j.jallcom.2024.175739
- J. Lin, L. Ma, Q. Liu, K. Xie, Y. Hu, L. Zhang, S. Li, M. Lu and G. Qiao, Continuous phase transition in thermoelectric Zn4Sb3, Mater. Today Energy, 2021, 21, 100787, DOI:10.1016/j.mtener.2021.100787
- W. Yang, Z. Guan, H. Wang, Y. Chen, H. Wang and J. Li, Ultrahigh anisotropic carrier mobility in ZnSb monolayers functionalized with halogen atoms, RSC Adv., 2022, 12(41), 26994–27001, 10.1039/D2RA04782A
- S. Malki, Z. Darhi, I. Guesmi, L. El Farh and A. Challioui, Transport Properties Study of ZnSb Compound Using BoltzTrap First-Principles, Mater. Sci. Forum, 2023, 1095, 3–9, DOI:10.4028/p-t4uPt3
- Z. Guan, W. Yang, H. Wang, H. Wang and J. Li, Direct band gap and anisotropic transport of ZnSb monolayers tuned by hydrogenation and strain, RSC Adv., 2022, 12(5), 2693–2700, 10.1039/D1RA08619G
- I. G. Elhoussieny, T. J. Rehaag and G. R. Bell, ZnSb Films on Flexible Substrates: Stability, Optical Bandgap, Electrical Properties, and Indium Doping, Adv. Electron. Mater., 2024, 10(3), 2300403, DOI:10.1002/aelm.202300403
- K. Zhou, T. Zhang, B. Liu and Y.-J. Yao, Electronic Structures and Thermoelectric Properties of ZnSb Doped with Cd and In from First Principles Calculations, Chin. Phys. Lett., 2020, 37(1), 017102, DOI:10.1088/0256-307X/37/1/017102
- S. Radha, J. Mani, A. B. S. Sunil Andrew, R. Rajkumar, M. Arivanandhan and G. Anbalagan, Inclusion of 2D nanosheets on ZnSb matrix as a unique approach to improve its thermoelectric performance, Mater. Sci. Semicond. Process., 2025, 186, 109031, DOI:10.1016/j.mssp.2024.109031
- A. Ostovari Moghaddam, A. Shokuhfar, Y. Zhang, T. Zhang, D. Cadavid, J. Arbiol and A. Cabot, Ge-Doped ZnSb/β-Zn4 Sb3 Nanocomposites with High Thermoelectric Performance, Defect and Dopant Mediated Thermoelectric Power Factor Tuning in β-Zn4Sb3, Adv. Mater. Interfaces, 2019, 6(18), 1900467, DOI:10.1002/admi.201900467
- V. Karthikeyan, T. Li, B. Medasani, C. Luo, D. Shi, J. C. K. Wong, K. Lam, F. C. C. Ling and V. A. L. Roy, Adv. Electron. Mater., 2020, 6(4), 1901284, DOI:10.1002/aelm.201901284
- D. Eklöf, A. Fischer, J. Grins, W. Scherer and U. Häussermann, Transport Properties of Ag-doped ZnSb, Z. Anorg. Allg. Chem., 2021, 647(2–3), 34–40, DOI:10.1002/zaac.202000314
- P. H. M. Böttger, G. S. Pomrehn and G. J. Snyder, Doping of p-type ZnSb: Single parabolic band model and impurity band conduction, Finstad, Phys. Status Solidi A, 2011, 208(12), 2753–2759, DOI:10.1002/pssa.201127211
- Z. Darhi, L. El Farh and R. Pandey, First-Principles Study of the Heterostructure, ZnSb Bilayer/h-BN Monolayer for Thermoelectric Applications, Materials, 2025, 18(2), 294, DOI:10.3390/ma18020294
- V. C. S. Theja, V. Karthikeyan, S. Nayak, K. U. Kandira, D. S. Assi, V. Kannan and V. A. L. Roy, Facile composite engineering to boost thermoelectric power conversion in ZnSb device, J. Phys. Chem. Solids, 2023, 178, 111329, DOI:10.1016/j.jpcs.2023.111329
- B. Fu, W. Wu, P. Zhang, H. Gu, X. Zhou, X. Zhu and J. Zhai, Impact of titanium modification on the performance improvement and phase change mechanism of ZnSb thin film, Surf. Interfaces, 2024, 53, 105044, DOI:10.1016/j.surfin.2024.105044
- Z. Song, W. Gu, Y. Ni and G. Wang, Period number tunable and thickness-dependent crystallization behavior of Sb/ZnSb-based superlattice-like phase change thin films, J. Alloys Compd., 2025, 1010, 178319, DOI:10.1016/j.jallcom.2024.178319
- W. Jia, Z. Cao, L. Wang, J. Fu, X. Chi, W. Gao and L.-W. Wang, The analysis of a plane wave pseudopotential density functional theory code on a GPU machine, Comput. Phys. Commun., 2013, 184(1), 9–18, DOI:10.1016/j.cpc.2012.08.002
- H. Chen, Y. Zhang, C. Zhou and Y. Zhou, Deep learning potential model of displacement damage in hafnium oxide ferroelectric films, npj Comput. Mater., 2024, 10(1), 270, DOI:10.1038/s41524-024-01465-6
- Y. Du, Z. Meng, Q. Yan, C. Wang, Y. Tian, W. Duan, S. Zhang and P. Lin, Deep potential for a face-centered cubic Cu system at finite temperatures, Phys. Chem. Chem. Phys., 2022, 24(30), 18361–18369, 10.1039/D2CP02758E
- D. Kussainova and Z. A. Panagiotopoulos, Simulation of lithium hydroxide decomposition using deep potential molecular dynamics, J. Chem. Phys., 2024, 161(13), 134506, DOI:10.1063/5.0230440
- M. S. Green, Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena, J. Chem. Phys., 1952, 20(8), 1281–1295, 10.1039/D2CP02758E
| This journal is © The Royal Society of Chemistry 2025 |